-
### In which tool?
gmx_MMPBSA_ana
### New Feature
Add tooltips to chart options
### Description
_No response_
### Relevance
medium
### Difficulty to implement
low
-
### My Question is...
is gmx_mmpbsa able to read input files from charmm-gui ? I have used amberf f14SB forcefield for protein and gaff2 for ligand, I tried to do calculations with protein 3htb wit…
-
**Describe the issue**
Hello,
Could you suggest what could be the problem for the below error message? and whether it affects the usage of gmx_MMPBSA program?
error message when gmx_MMPBSA is cal…
-
- [x] Add a flag to overwrite or not the folder where the repository with the examples was cloned.
It would be something like `gmx_MMPBSA_test` [`-nr` / `-noreuse`] (Default = True)
`-nr` wi…
-
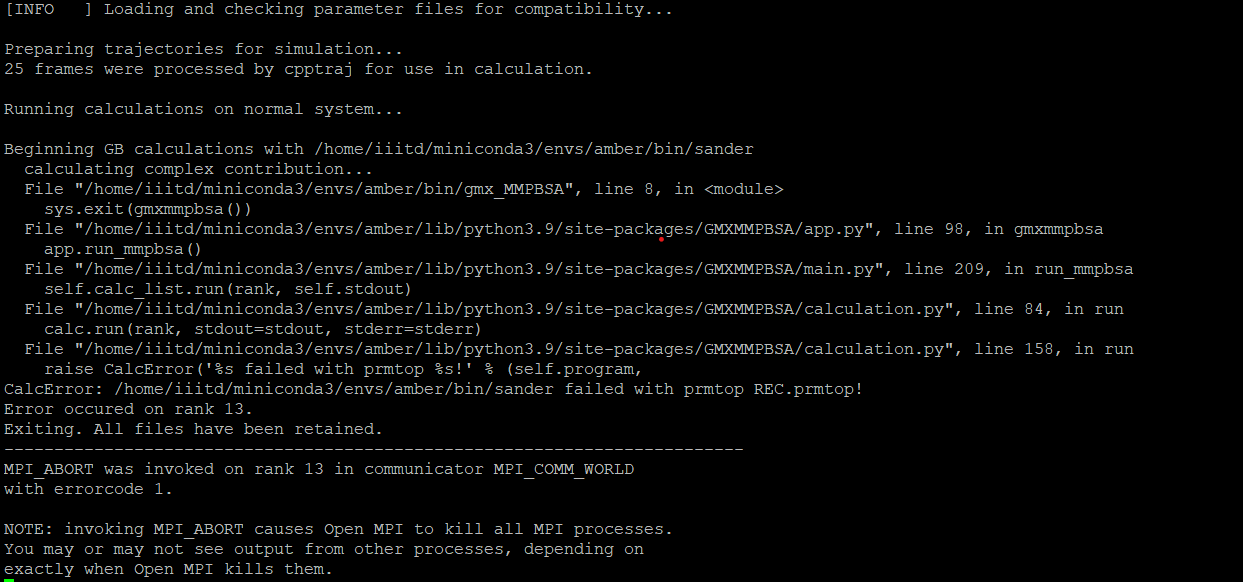
I have encountered this error whil running gmx_MMPBSA.
The command used was
gmx_…
-
**Describe the issue**
```
"""
Traceback (most recent call last):
File "/home/x/software/anaconda3/envs/amber/lib/python3.9/multiprocessing/pool.py", line 125, in worker
result = (True, fun…
-
Hi, everyone. Sorry for the naive question. I'm trying to use gmx_MMPBSA on a linux machine (gmx_MMPBSA v1.4.3 based on MMPBSA version 16.0 and AmberTools 20) with a protein-ligand complex using gromo…
-
Hello Sir,
I have encountered the below error when I try to run gmx_MMPBSA_test. Could you suggest a way to fix it?
I used ubuntu 18.04 and have python3.6.9 installed in the system.
#The error…
-
### In which tool?
gmx_MMPBSA
### New Feature
Improvement of "print_res" function for &decomp and QM
- if invalid selection, apply dynamic selection scheme (distance increase)
- improve doc…
-
### Bug summary
I've broken more things! When using the MT approach, I put my topologies in separate folders for complex, receptor and ligand (named as complex, receptor, ligand, with .top files to…