-
Hi Adrian,
Using an aligner such as STAR or Tophat seems to give more alignments/content from indrop sequence reads compared to the current Bowtie aligner usage. Would it be possible to incorporate…
-
Key points for introduction:
- What are genome annotations? What type of features are included in genome annotations (protein-coding genes, pseudogenes, RNAs etc)? What sorts of things do biologists …
-
BGC0002687
This entry is missing a central gene identified in the paper but seems missing for the MiBIG entry. It also doesn't appear to be recognised if you run the assembled genome through antiSM…
-
https://www.ncbi.nlm.nih.gov/IEB/ToolBox/CPP_DOC/doxyhtml/asnval_8cpp_source.html
The problem is that this is C++ and we have to convert these to ASN. Distributing the C++ binaries prebuilt might …
-
Hi Seeman,
I downloaded few plasmids sequences in Genbank full format from NCBI for plasmid annotation and ran the following command:
`prokka ENT2_Contig6_len_41186_circ_NDM-1_Plasmid.fasta --ou…
-
Version: 0.5.2
Date: 2018-07-05
> library(HGNChelper)
> new.hgnc.table
-
Currently, after the [biotype cleanup](https://github.com/hammerlab/pyensembl/pull/165), only the biotype "protein_coding" is used in the check in [`Transcript.is_protein_coding()`](https://github.com…
joaoe updated
8 years ago
-
Hello,
Not sure who is monitoring these issues now @satta has left Sanger, but I wasn't sure where else to send it.
Sascha showed me how to run companion on the command line using a fungidb refe…
-
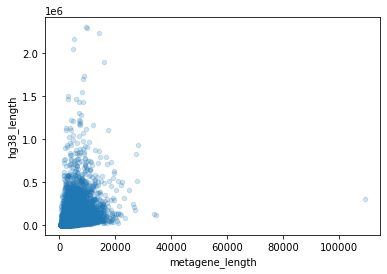
I computed gene lengths using end-start from the hg38 gtf. Then I compared to the sum …
-
Hi,
I wonder if SIFT can predict synonymous deleterious? I found some novol mutation didn't affect the aa, but with SIFT score