 duypham2108
commented
1 year ago
duypham2108
commented
1 year ago Can you write the anndata object and send me?
adata.write_h5ad("adata_object.h5ad")Anyways, it's holiday now but I will check it ASAP.
Closed bmlett closed 1 year ago
duypham2108
commented
1 year ago Can you write the anndata object and send me?
adata.write_h5ad("adata_object.h5ad")Anyways, it's holiday now but I will check it ASAP.
 bmlett
commented
1 year ago
bmlett
commented
1 year ago Hi,
Commands used to get A1_count_matrix.csv and A1_spatialCoords.csv
count_matrix = t(counts(data)) spCoords = as.data.frame(spatialCoords(data)) array_coords = colData(data)[2:3] spatial = cbind(spCoords, array_coords) names(spatial)[1:2] = c("imagecol", "imagerow")
These are the commands run to import the data.
count_matrix = pd.read_csv("A1_count_matrix.csv") xy = pd.read_csv("A1_spatialCoords.csv") adata = st.create_stlearn(count=count_matrix,spatial=xy,library_id="A48_A1", image_path="tif/WSA_LngSP10193345.tif",scale=1,background_color="white")
The adata.write_h5ad("adata_object.h5ad", compression="gzip") result is: [https://res-geo.cdn.office.net/assets/mail/file-icon/png/generic_16x16.png] adata_object.h5adhttps://uwprod-my.sharepoint.com/:u:/g/personal/blett_wisc_edu/Eajcx9wAG4FHgjbDuy4sflwBfDAeUdV7p4R6Lgq1XsLALw
By adding the following after building the stlearn object, it seems to fix the issue.
adata.obs['array_row']=xy.iloc[:,2] adata.obs['array_col']=xy.iloc[:,3]
Though if there is a better way to import the data from R SpatialExperiment object that would be advantageous to learn about.
Thanks and happy holidays!
Beth M. Lett, Ph.D. Postdoctoral Trainee fellow - ERP | Ong Lab School of Medicine and Public health (SMPH), ADMIN, & Endocrinology and Reproductive Physiology (ERP) Pronouns: She, Her, Hers Office Address: 2778 WIMR West 1111 Highland Ave Madison, WI 53705
From: Duy Pham @.> Sent: Thursday, December 29, 2022 7:56 PM To: BiomedicalMachineLearning/stLearn @.> Cc: BETH LETT @.>; Author @.> Subject: Re: [BiomedicalMachineLearning/stLearn] Error: "array_row" not found during SME (Issue #219)
Can you write the anndata object and send me?
adata.write_h5ad("adata_object.h5ad")
Anyways, it's holiday now but I will check it ASAP.
— Reply to this email directly, view it on GitHubhttps://github.com/BiomedicalMachineLearning/stLearn/issues/219#issuecomment-1367680911, or unsubscribehttps://github.com/notifications/unsubscribe-auth/AH4C24W2XNZ72HK3CPJ5RDTWPY6ODANCNFSM6AAAAAATLPDXRE. You are receiving this because you authored the thread.Message ID: @.***>
duypham2108
commented
1 year ago Good to know this issue is solved. We will try to make a function to convert from R objects like SpatialExperiment or SeuratObject in the near future. Thanks for suggestion
bmlett
commented
1 year ago There seems to be additional issues with using the create.stlearn for downstream functions. When trying to run the st.spatial.trajectory.pseudotime. When I run it on the above data it hits an error:
Traceback (most recent call last): File "<stdin>", line 1, in <module> File "/ua/blett/anaconda3/envs/stlearn/lib/python3.8/site-packages/stlearn/spatials/trajectory/pseudotime.py", line 158, in pseudotime adata.uns["global_graph"]["graph"] = nx.to_scipy_sparse_array(G) File "/ua/blett/anaconda3/envs/stlearn/lib/python3.8/site-packages/networkx/convert_matrix.py", line 880, in to_scipy_sparse_array raise nx.NetworkXError("Graph has no nodes or edges") Graph has no nodes or edges
However when I use Read10x this error does not happen. I compared the two anndata objects and noticed it stores different values in the .uns['spatial'] section. I am not sure of a good way to fix this as there are qc steps in R I wish to run before preforming clustering. Thanks for developing this tool. I am hoping something comes from this thread soon: https://github.com/theislab/zellkonverter/issues/61 to create a nice way to convert between similar to singlecell.
duypham2108
commented
1 year ago In this step, there is a parameter to define the distance between every adjacent nodes: eps (based on DBSCAN). The Visium data is ~2000x2000 px then we use eps = 50 as default here. But it will depend on your data like what is the average distance between every adjacent nodes? Then you can use that distance to specify the eps parameter here:
st.spatial.trajectory.pseudotime(data,eps=50,use_rep="X_pca",use_label="louvain")Hi -
Thanks for the reply. I tried various values in the eps (2,18,50,100) and all values returned the same error for the data load using create.stlearn. I even tried two different 10x genomic datasets. When I switched to the Read10x method used in the trajectory example that worked.
I looked at the one 10x genomics dataset annData when load using create.stlearn and Read10x. This is the first few lines of the spatial adata.uns file between the two loading methods.
create.stlearn
OverloadedDict, wrapping: OrderedDict([( 'spatial', {'Bcancer_A1': {'images': { 'hires': array([[[188, 192, 191], [188, 192, 190], [188, 191, 188],
Read10x
OverloadedDict, wrapping: OrderedDict([( 'spatial', {'Parent_Visium_Human_BreastCancer': {'images': {'hires': array([[[0.7294118 , 0.74509805, 0.7372549 ], [0.7294118 , 0.74509805, 0.7372549 ],
The other key difference is in the scalefactors:
create.stlearn
'use_quality': 'hires', 'scalefactors': {'tissue_hires_scalef': 1, 'spot_diameter_fullres': 50}}})])
Read10x
'scalefactors': {'spot_diameter_fullres': 177.4984743134119, 'tissue_hires_scalef': 0.08250825, 'fiducial_diameter_fullres': 286.7283046601269, 'tissue_lowres_scalef': 0.024752475},
It makes sense to me that the Read10x method would have more information since the create.stlearn is only provided a base amount information. Does the fact that the create.stlearn version being whole numbers imply that the eps value needs to be higher?
duypham2108
commented
1 year ago Basically, the difference is about the spatial information scale. The Visium data provided all the informatio like scalefactors. In the Read10X function, we store the raw spatial info in adata.obsm["spatial"] and adata.obs[["imagecol","imagerow"]] which is the .obsm["spatial"] * tissue_hires_scalef for example of the hires image. In the create.stlearn, we only store raw spatial info and you willl see both adata.obsm["spatial"] and adata.obs[["imagecol","imagerow"]] are similar with the scale factor = 0.
In downstream analysis, we use those spatial information to construct the neighborhood array for each spot/cell and also the input for the local clustering (using spatial data only) by using DBSCAN. The eps parameter defines how separate the cluster should be. Then it will depends on your spatial information scale as the input. I would say that you should look on your spatial information scale to set the eps value. Another way is using parameter scale in create_stlearn function. For example, set it with max(your_spatial_coordinate) / 2000 and then use similar eps with the tutorial.
bmlett
commented
1 year ago Thank you for that explanation!! By adding the scale information during creation, I was able to get the data to run throughst.spatial.trajectory.pseudotime. Thank you for the patience and explaining the difference in the two methods. I do have one final follow up question regarding the spot_diameter_fullres and what the purpose is for this argument? This is partially out of curiosity and partially because though I provided an argument for the spot_diameter_fullres in create.stlearn viewing the data.uns shows the value as 50.
Again, thank you so much for answering my questions!!
duypham2108
commented
1 year ago It will be used in the CCI prediction part if you want to calculate the distance for neighborhood spots automatically
https://github.com/BiomedicalMachineLearning/stLearn/blob/ebc9b52f2b4adf7753e264af22ffa9aaa4e175d1/stlearn/tools/microenv/cci/base.py#L64 Otherwise, it's not so important. Also, it only be useful when you use the Visium or any platform that have constant distance between spots. Hope it helps
{kind=link}
Good morning. I get the below error when attempting to run the st.spatial.SME.SME_normalize command.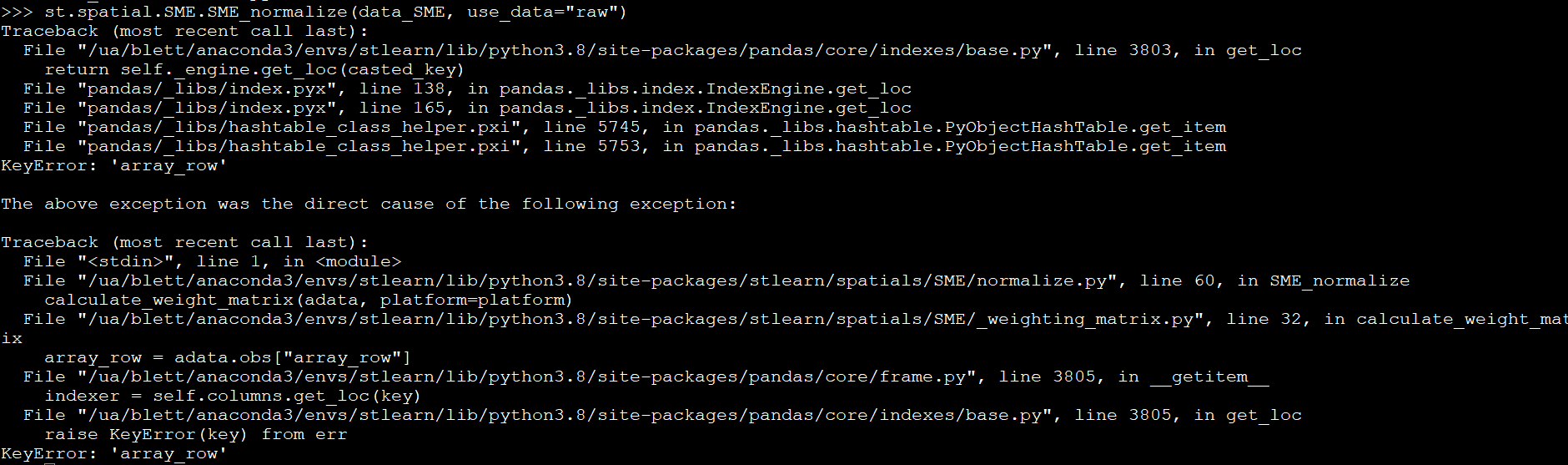
I used create_stlearn to import data where the counts_matrix is from a spatial experiment object counts(spe) command and the spatial data is from as.data.frame(spatialCoords(spe)). I even attempted to add the array_col and array_row data from my SpatialExperiment obje to the spatialcoordinates file assuming this was the issue and still obtain the above error.
Thanks