 aryarm
commented
2 years ago
aryarm
commented
2 years ago Just recording a suggestion from @RossDeVito for us to use multi-processing!
from functools import partial
import numpy as np
from multiprocessing import Pool
def single_check(idxs, arr):
return np.all(arr[:, idxs], axis=1)
def parallel_loop(idxs, arr, num_haps, n_processes=None):
with Pool(processes=n_processes) as pool:
res = pool.map(partial(single_check, arr=arr), idxs)
return np.stack(res, axis=1)I was initially hesitant to use this because the transform command itself will be run in a multi-threaded way: we expect it to be called multiple times in parallel for each chromosome. But perhaps the multi-threading could just be an option for the user. Another consideration is that the arrays might be getting copied when we use subprocesses, which itself might take some time.
In any case, I would still like to explore other options to improve the speed first. I know that we'll ultimately have to use multi-processing in the end, but I'd like to leave it as the method of last resort, especially since other strategies we implement may change everything anyway? This code will definitely be helpful to have for then.
 d-laub
d-laub
Description
Transforming a set of haplotypes can take a while, especially if you have many haplotypes. A quick benchmark via the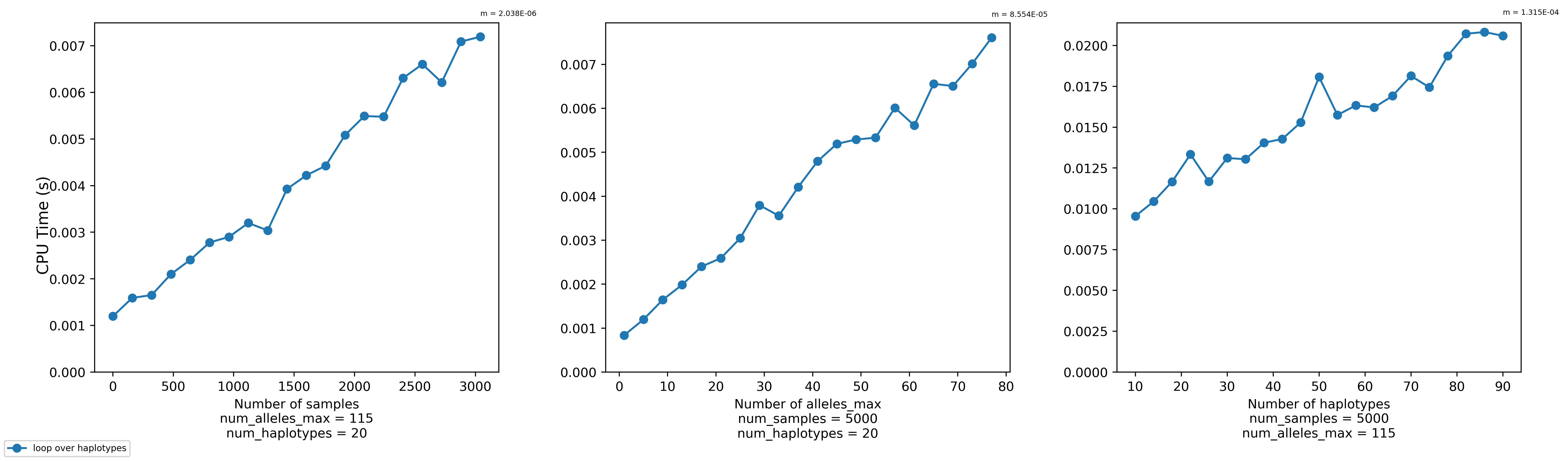
bench_transform.pyscript seems to indicate that it scales linearly with the number of samples, the max number of alleles in each haplotype, and the number of haplotypes.@s041629 wants to transform roughly 21,000 haplotypes for 500,000 samples where the max number of alleles in a haplotype is 115. Currently (as of f45742f466ac0eb1de6d0358494d6ace8fd5856f), the
Haplotypes.transform()function takes about 10-25 real minutes to accomplish this task. (It varies based on the willingness of the kernel to prioritize our job on the node.)For now, I think this will be fast enough. But we might want to brainstorm some ideas for the future. In any case, I don't really think I can justify spending a lot more time to improve this right now (since I've already spent quite a bit), so I'm gonna take a break and solicit suggestions until I feel energized enough to tackle this again.
Details
The vast majority of the time seems to be spent in this for-loop.
https://github.com/CAST-genomics/haptools/blob/639985ec639b0f45be36f7f3e6641152dc056267/haptools/data/haplotypes.py#L1054-L1055
Unfortunately, I can't use broadcasting to speed up this loop because the size of
idxs[i]might vary on each iteration. And I can't pad the array to make broadcasting work because I quickly run out of memory for arrays of that shape. Ideally, I would like to find a solution that doesn't require multi-threading or the installation of new dependencies besides those we have already.Steps to reproduce
A basic test
This test uses just 5 samples, 4 variants, and 3 haplotypes. It's a sanity check to make sure the
Haplotypes.transform()function works and that none of the code will fail.A benchmarking test
To benchmark the function and see how it scales with the number of samples, max number of alleles in each haplotype, and the number of haplotypes in each allele, you can use the
bench_transform.pyscript as so. It will create a plot like the one I've included above.https://github.com/CAST-genomics/haptools/blob/639985ec639b0f45be36f7f3e6641152dc056267/tests/bench_transform.py#L19-L21
A pure numpy-based test of the for-loop
The tests above require that you install the dev setup for
haptools. If you'd like to reproduce just the problematic for-loop for @s041629's situation in pure numpy you can do something like this:A full, long-running test
If requested, I can provide files that reproduce the entire setup so that you can execute the
transformcommand with them. The files are a bit too large to attach here.Some things I've already tried
arr(akaequality_arr) of size equal to the number of haplotypes and then performingnp.all()on the other dimension. This takes longer and requires too much more memory. See fac79b07c085490351b1ad2e3b61bc06732d731b.num_alleles < num_haps, at least in this situation). This was a clever strategy from Ryan Eveloff. Strangely, this also took longer than looping over the haplotypes. I'm not sure why. See 335e5f7063f84f8f739dd62e789cd5a48a1468df.Potential ideas
.hapfile Matteo gave me indicates that there would be at least 1,000 leaves in such a tree.