 sjackman
commented
9 years ago
sjackman
commented
9 years ago - ABySS 1.9.0
Closed sjackman closed 9 years ago
sjackman
commented
9 years ago  jts
commented
9 years ago
jts
commented
9 years ago I implemented GFA in a fork of daligner: https://github.com/jts/daligner
sjackman
commented
9 years ago That's amazing! I had just said to @pmelsted that I thought GFA could/should be the format for long read overlap alignments. @JustinChu
sjackman
commented
9 years ago @jts I'm curious though, why fork and patch DALIGNER rather than create a DALIGNER format to GFA format conversion script? Or was there insufficient information stored in the DALIGNER output?
jts
commented
9 years ago The latter. The DALIGNER output files don't store full alignments, only anchors that can rebuild the alignment on demand.
sjackman
commented
9 years ago Ah. Was the modification a new SAM GFA output format to LAshow, then? Any thoughts of submitting a pull request upstream? It seems like a very useful feature to me.
 pmelsted
commented
9 years ago sjackman
commented
9 years ago
pmelsted
commented
9 years ago sjackman
commented
9 years ago Cool. Would you add them to https://github.com/pmelsted/GFA-spec/blob/master/README.md#implementations ?
sjackman
commented
9 years ago @jts I got my wires crossed above doing too many things at once. Did you add a GFA output format to LAshow?
 ekg
commented
9 years ago
ekg
commented
9 years ago @pmelsted good to see that you have a (GFA-producing) tool too.
So I need to implement that 1-character change to bring myself into concordance with the other implementations.
ekg
commented
9 years ago vg needs to feed something back into the spec--- Paths! I will start a separate thread about this.
jts
commented
9 years ago @sjackman it is a convertor from .las to .gfa: https://github.com/jts/DALIGNER/blob/master/LA2gfa.c
sjackman
commented
9 years ago So I need to implement that 1-character change to bring myself into concordance with the other implementations.
@ekg What's the change? Just curious.
sjackman
commented
9 years ago @ekg @vg If you add file paths, be sure to add SHA256 for those files as well, to ensure coherency.
sjackman
commented
9 years ago @jts Any idea what's going on here? LAshow works fine on the same file. Would you consider changing the command line format of LA2gfa to be the same as LAshow for consistency? Namely, making the db parameter the first positional argument rather than -a option.
❯❯❯ LA2gfa -a:pg29mt-scaffolds.db pg29mt-scaffolds.las
H VN:Z:1.0
LA2gfa: Index out of bounds (Load_Read)
❯❯❯ LAshow -o pg29mt-scaffolds.db pg29mt-scaffolds.las |wc
8 111 649My edge orientations aren't written the same as other tools. I show L 1 - 2
So I need to implement that 1-character change to bring myself into concordance with the other implementations.
What's the change? Just curious.
— Reply to this email directly or view it on GitHub https://github.com/pmelsted/GFA-spec/issues/3#issuecomment-124183498.
 lh3
commented
9 years ago
lh3
commented
9 years ago This makes me want to change this "+/-" thing. What about L 1 3 2 5, meaning the 3'-end of 1 joins/overlaps the 5'-end of 2? Maybe too ugly and/or too late?
ekg
commented
9 years ago I'd prefer not to use 3 and 5 for the ends of the nodes. Better to avoid likely node identifiers.
But then I assumed that the link was from the plus end to minus end. Actually the + and - indicate the edges direction relative to the node. Is that right?
I'm concerned there will be problems representing things like links from the end of a node to the start. Maybe I am not understanding still. On Jul 23, 2015 8:13 PM, "Heng Li" notifications@github.com wrote:
This makes me want to change this "+/-" thing. What about L 1 3 2 5, meaning the 3'-end of 1 joins/overlaps the 5'-end of 2? Maybe too ugly and/or too late?
— Reply to this email directly or view it on GitHub https://github.com/pmelsted/GFA-spec/issues/3#issuecomment-124191718.
pmelsted
commented
9 years ago Yes + and - refer to whether you should use the nucl. sequence of the segment as presented in the S line or (-) the reverse complement.
(stealing this from @jts LA2gfa.c)
//
// GFA link orientations:
//
// S1 -------------->
// S2 --------------->
// Record: S1 + S2 +
// S1 -------------->
// S2 ------------->
// Record: S2 + S1 +
// S1 -------------->
// S2 <---------------
// Record: S1 + S2 -
// S1 <--------------
// S2 -------------->
// Record: S2 + S2 -I'm also open to using ~ for reverse complement (this was in fastg) but I like that both orientations + and - have to be specified.
ekg
commented
9 years ago Wait, is there any way to have a link from the 5' end of a node to the 5' end of another node?
If not then GFA will be incompatible with what the GA4GH group is producing. There is a lot of pressure to represent inversions natively in the reference graphs. On Jul 23, 2015 8:20 PM, "Pall Melsted" notifications@github.com wrote:
Yes + and - refer to whether you should use the nucl. sequence of the segment as presented in the S line or (-) the reverse complement.
(stealing this from @jts https://github.com/jts LA2gfa.c)
// // GFA link orientations: // // S1 --------------> // S2 ---------------> // Record: S1 + S2 +
// S1 --------------> // S2 -------------> // Record: S2 + S1 + // S1 --------------> // S2 <--------------- // Record: S1 + S2 - // S1 <-------------- // S2 --------------> // Record: S2 + S2 -I'm also open to using ~ for reverse complement (this was in fastg) but I like that both orientations + and - have to be specified.
— Reply to this email directly or view it on GitHub https://github.com/pmelsted/GFA-spec/issues/3#issuecomment-124194068.
lh3
commented
9 years ago is there any way to have a link from the 5' end of a node to the 5' end of another node?
That is L 1 - 2 +.
There are at least two contradicting ways to interpret this +/-. @ekg, Richard Durbin and a few others are using the other interpretation. It is more straightforward to say the beginning of segment 1 joins the end of segment 2.
lh3
commented
9 years ago using ~ for reverse complement (this was in fastg)
Thought about that, but this is awkward to awk, as you can't find a node with awk '$2=="abc"'.
sjackman
commented
9 years ago I prefer to view the overlaps as alignments. Some portion of sequence A aligns to some portion of sequence B. That portion of A and B is either reverse complemented - or not +. That's what the + and - operators indicate. This is very similar to the exonerate --showcigar format, which I think is quite nice:
cigar: 5 290800 308816 + 12 0 18016 + 90062 M 18016
A portion of 5 + overlaps a portion of 12 +.
pmelsted
commented
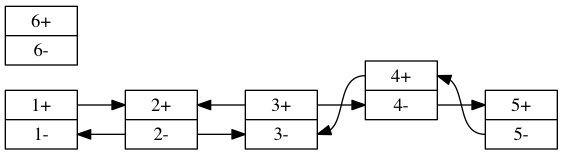
9 years ago Also note that links are contravariant (probably not the right term, but works) , A + B + is equivalent to B - A - and A - B + is equivalent to B - A +
One way to visualize the links is to draw two nodes per segment a + and - version and connect them appropriately.
jts
commented
9 years ago @sjackman LA2gfa is very out of date so I suspect something in the underlying files changed
sjackman
commented
9 years ago Yes, I believe the database did change at one point. https://github.com/thegenemyers/DAZZ_DB/blob/master/README#L40-L47
UPGRADE & DEVELOPER NOTES ! ! ! If you have already built a big database and don't want to rebuild it, but do want to use a more recent version of the software that entails a change to the data structures (currently the updates on Sept 25, 2014 and December 31, 2014), please note the routines DBupgrade.Sep.25.2014 and DBupgrade.Dec.31.2014.
pmelsted
commented
9 years ago @ekg Regarding 5' to 5' links. This is not possible right now and I think we should talk about it in another issue. One way to represent it would be to have a separated record for the inverted segment.
ekg
commented
9 years ago This would be easy to represent if the links were defined as between ends rather than as relative to the natural orientation of the nodes. Would we lose anything relative to the current representation by changing these semantics?
sjackman
commented
9 years ago 5' to 5' links are supported in the current spec. The following snippet connects the leftmost end of 0 to the leftmost end of 1, AAA to GGG.
S 0 AAA
S 1 CCC
L 0 + 1 - 0MAnd a path:
P 2 0+,1- 0MNo, @sjackman what @ekg is referring to is if you have an inversion, like
S 0 GAT
S 1 TAC
S 2 A
L 0 + 1 + 0M
L 1 + 2 + 0M
P 3 0+,1+,2+, 0M,0MBut what if the TAG was inverted how would you encode the links and path for GAT-CAT-A rather than GAT-TAC-A. Currently this can't be done without introducing the segment CAT and the appropriate links.
sjackman
commented
9 years ago When you say inverted, do you mean reversed without being complemented? Does that happen? How would that happen?
 pb-jchin
commented
9 years ago
pb-jchin
commented
9 years ago I am a little bit confused by the GFA implemention of the daligner. From the assembly pipeline point of view, daligner's output is the overlapping information, not the assembly graph. Maybe @jts can help me understand why we use GFA to store overlapping. Thanks a lot.
The FALCON assembler that I implemented dose convert the overlap ouput as some explicitly overlap information (as a small variation of the blasr -m4 output.
The graph representation used in FALCON is read-end based. It is more like the 1st version Heng Li proposed, but I don't explicity store the sequence. The sequences can be inferred from the read end labelling and the original sequences. There is pros and cons to store seqeunces with the linkage information. Currently, it is easier for me to code to store them separately. It makes the I/O for graph analysis little bit eaiser without reading the seqeunce data.
jts
commented
9 years ago @pb-jchin one of the original goals of GFA (and SQG, iirc) was that it could act as an intermediate format for the entire assembly pipeline from input reads to an output graph. We could then write a pipeline of standalone software with GFA input/output for each stage.
pb-jchin
commented
9 years ago @jts thanks for the clarification. Currently, for PacBio related assembly work, we basically seperate the sequences itself and the metadata into different files. For example, the daligner db file and las file. I use the same for downstream too. (I think CA/WGS have a couple different storages too.) Just wondering what's people's thought about such sperations. I am not sure what is the working draft for the curren GFA format. The linkage information and the sequence information are in different lines. It may be a good idea if they are in different files. (or bad idea, perhaps)
pb-jchin
commented
9 years ago @jts maybe we are acutally talking about common format for storing overlaping data rather than the final assembly graph?
sjackman
commented
9 years ago @pb-jchin
Currently, for PacBio related assembly work, we basically seperate the sequences itself and the metadata into different files. … Just wondering what's people's thought about such separations.
I'm in favour of storing the sequences separately from the graph information. I'm not a fan of duplicating information. You may put * in the sequence column of the S lines, and you can use the LN attribute to specify the sequence length. ABySS does this. It puts the contig sequences in an indexed FASTA file, and the overlap information in a GFA or GraphViz file.
ekg
commented
9 years ago From my perspective an ideal non-redundant representation would describe a sequence graph with S and L entries, and then use path or containment records to enumerate input reads/contigs as walks through this graph.
This would also allow merging the cigars of the links into the graph itself, which would simplify parsing the graph. The cigars are themselves like a mini graph format.
sjackman
commented
9 years ago I believe the current GFA graph format can be used to describe such a string graph. The CIGAR strings can all be 0M in that case. It can also be used to describe a sequence overlap graph, which is a use case that's very useful to long-read-overlaps and contig overlap graphs.
sjackman
commented
9 years ago If anyone finds additional implementations of GFA, please open a pull request to add it/them to the README.md.