 cmdcolin
commented
10 years ago
cmdcolin
commented
10 years ago Do you have a link for how this looks? I don't think we have this feature
http://www.broadinstitute.org/igv/view_as_pairs
You could however, color by template length, which might give you some ideas for how the paired end reads are linked in a region.
You can do this by using the style.color as a javascript callback (short mailing list thread here with example http://sourceforge.net/p/gmod/mailman/message/32619454/)
Other links http://gmod.827538.n3.nabble.com/Gmod-ajax-connecting-paired-end-reads-with-Alignments2-td4045949.html
 selewis
selewis Kinztan
Kinztan rbuels
rbuels scottcain
scottcain keiranmraine
keiranmraine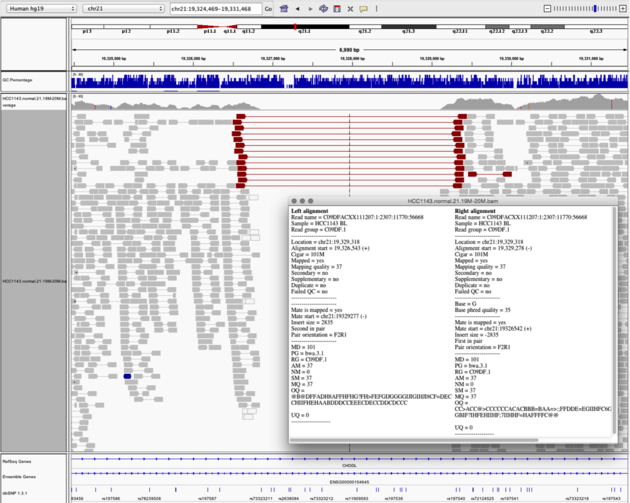

 malonge
malonge
Is there any function that can enable to visualize the paired end links between two pair ends. Just like the broad IGV did, it is quite important to visualize the linkage of pair end sequence to understand their mechanism. Is there any configuration I need to change for that?