 pbuslaev
commented
4 years ago
pbuslaev
commented
4 years ago I have the code to calculate the distributions and I will do that soon. I will also check again the snapshots for head group conformations. I also can add the possibility to calculate P-N vector angle to the code. Will comment about all that soon



 markussmiettinen
markussmiettinen ohsOllila
ohsOllila





 hsantila
hsantila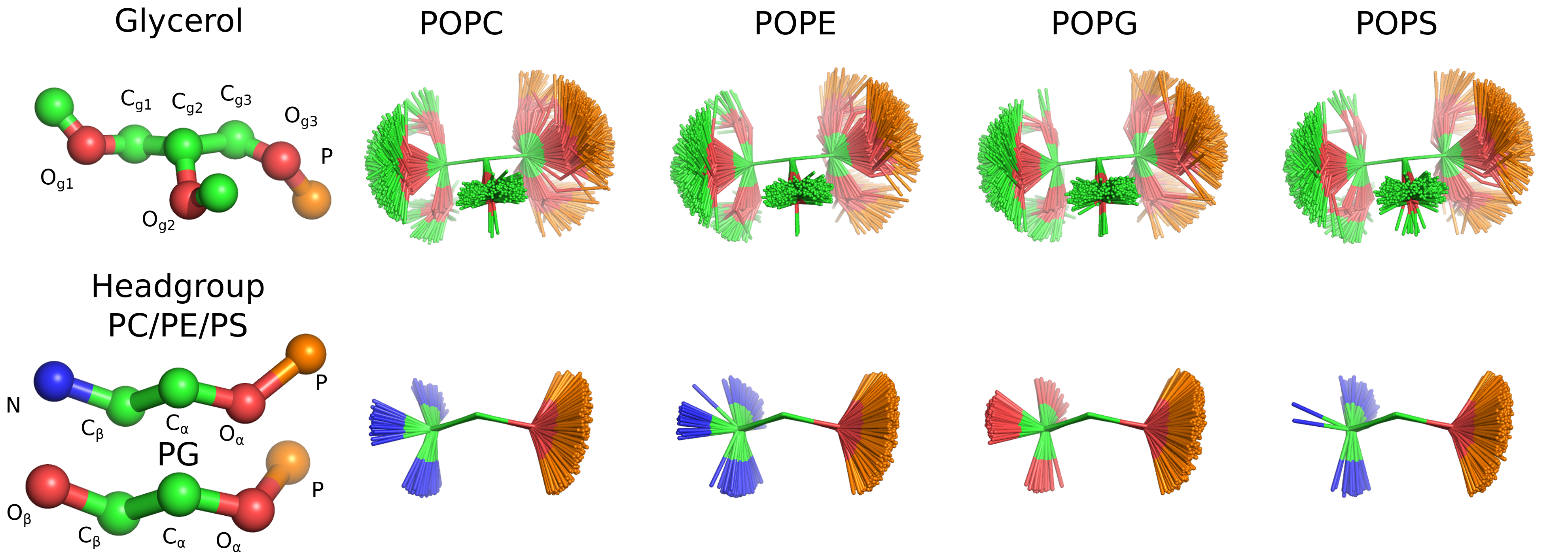{kind=link}
As discussed in the manuscript, CHARMM36 reproduces the essential differences in order parameters between PC, PE, PG and PS lipids. Therefore, it is reasonable to analyze the structural differences between headgroups from CHARMM36 simulations.
We already have a visualization of snapshots, but at least dihedral distributions are needed.
Also, the analysis of P-N angle with respect to membrane normal could be useful.
Also, more ideas on useful and illustrative quantities to compare between headgroups are welcomed.