 wangwei1619
commented
5 years ago
wangwei1619
commented
5 years ago Maybe I can share some development by myself after several days of trying.
In Events building block in MoBi, I changed the value of Use as Suspension to 0, resulting in the parameter Tablet_Location increasing druing simulation. Each Tablet_Location value is responsible for just one GI segment where drug release takes place as if drug would be dissolved in different segments at different time.
^^^^
However, simulation result get worse even though I think this change is an improvement.

 Yuri05
Yuri05

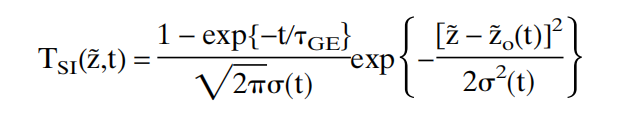
 with the [1-fabs(z,t)] replaced by the dissolution process selected (Weibull / Table / …)?
with the [1-fabs(z,t)] replaced by the dissolution process selected (Weibull / Table / …)? teutonicod
teutonicod The C_max is almost double of that from Poulin & Theil model, and the t_amx is much advanced.
I think the potential reason is the fu of carvedilol is 0.0054, very small, so that its concentration should be infunced by protein constituent. PK-Sim standard distribution model takes account the protein as a separate compart, so its simulation is more accurate in this case.
However, when I compare these two simulations in MoBi, I find the Partition coefficient (blood cells/plasma) defined in molecule in both simulations is equal with the same formula taking protein into the account. Besides, in organism, there are Vf(water, plasma) and Vf(water, plasma)-P-T, defining the fractional volume content of water in plasma used in PK-Sim standard and Poulin & Theil model respectively, but in somewhere, e.g. partition coefficient (interstial/plasma), only Vf(water, plasma) is used in both models. So, where is the difference between these two models embodied actually in PK-Sim or MoBi?
The C_max is almost double of that from Poulin & Theil model, and the t_amx is much advanced.
I think the potential reason is the fu of carvedilol is 0.0054, very small, so that its concentration should be infunced by protein constituent. PK-Sim standard distribution model takes account the protein as a separate compart, so its simulation is more accurate in this case.
However, when I compare these two simulations in MoBi, I find the Partition coefficient (blood cells/plasma) defined in molecule in both simulations is equal with the same formula taking protein into the account. Besides, in organism, there are Vf(water, plasma) and Vf(water, plasma)-P-T, defining the fractional volume content of water in plasma used in PK-Sim standard and Poulin & Theil model respectively, but in somewhere, e.g. partition coefficient (interstial/plasma), only Vf(water, plasma) is used in both models. So, where is the difference between these two models embodied actually in PK-Sim or MoBi?
Hi, all
I am following a simulation in one paper (Manuel Ibarra, Cristian Valiante, Patricia Sopeña, et al. Integration of in vitro biorelevant dissolution and in silico PBPK model of carvedilol to predict bioequivalence of oral drug products. European Journal of Pharmaceutical Sciences, 2018,118: 176-182). In this paper, author input the Formulation as a Table of an in vitro dissolution result which mimics the time and pH when drug goes through GI tract like this :
I define the Administration Protocol as User Defined with Target organ as stomach , Target compartment as plasma and I also use the same dissolution tale in formulation. Finally I get a similar but moderately different result in plasma concentration profile like this: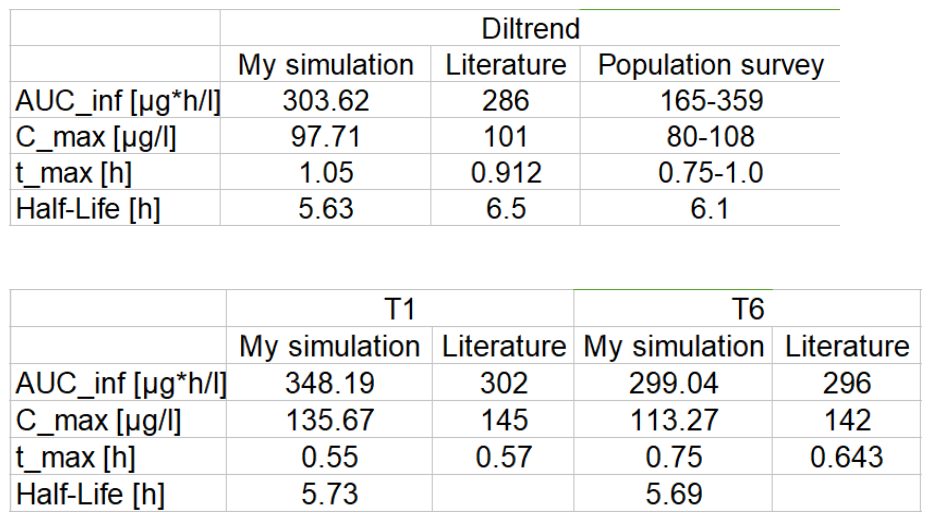
I want to ask if there is any way more acceptable to simulate these data?