 prvmalik
commented
5 years ago
prvmalik
commented
5 years ago Try optimizing intestinal permeability (transcellular) in the parameter identification tool.
With pKa 7.1 a notable portion of your molecule will be charged at physiologic pH. Try switching the cell permeability calculation algorithm to charge-dependent schmitt. Of course this will change your blood:plasma ratio.
On opinion alone, I find that using a dummy enzyme inside hepatic intracellular space works better than using the total hepatic plasma clearance function, especially in the case when permeability may be limiting to elimination.
And of course, this drug is a major substrate for transporters. You'll have to read into the literature to find which ones and to which degrees the drug maybe transported. Try using literature-based Km for PgP and then optimizing Vmax according to the fraction excreted unchanged in bile/feces.
 wangwei1619
wangwei1619 I want to see where the drug accumulates in and I find concentration curve for every tissue is showing decreasing shape. But for small intestine and the intracellular space of duodenum, jejunum and ileum, the Cmax are especially high.
I want to see where the drug accumulates in and I find concentration curve for every tissue is showing decreasing shape. But for small intestine and the intracellular space of duodenum, jejunum and ileum, the Cmax are especially high.
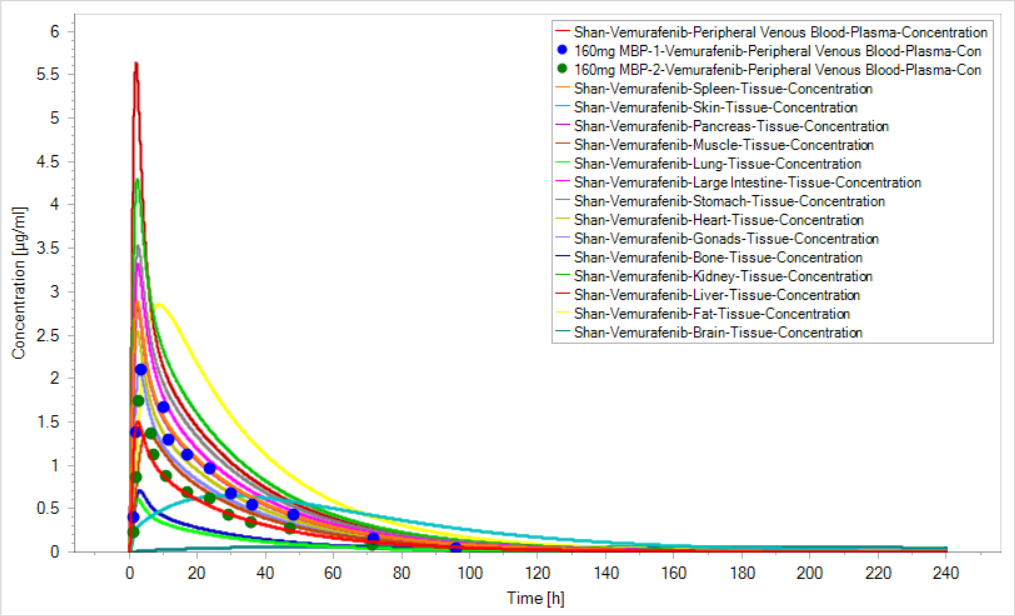
 How can drug concentration in every tissue decrease when the clearance is 0?
And, I know there is a mechanism of scaling the permeability between intracellular and interstial space in mucosa to avoid drug accumulation in enterocytes, does the figure mean an accumulation in my simulation (I use the specific intestinal permeability and organ permeability calculated automatically )?
How can drug concentration in every tissue decrease when the clearance is 0?
And, I know there is a mechanism of scaling the permeability between intracellular and interstial space in mucosa to avoid drug accumulation in enterocytes, does the figure mean an accumulation in my simulation (I use the specific intestinal permeability and organ permeability calculated automatically )?  tobiasK2001
tobiasK2001 StephanSchaller
StephanSchaller



 Thanks for your help, I will folllow your guide .
Thanks for your help, I will folllow your guide . I only define one individual and one compound, and associate the vemurafenib to the transporters, but I transporters are not showed in simulation.
Is there anyway to solve this?
I only define one individual and one compound, and associate the vemurafenib to the transporters, but I transporters are not showed in simulation.
Is there anyway to solve this? msevestre
msevestre leeking-55
leeking-55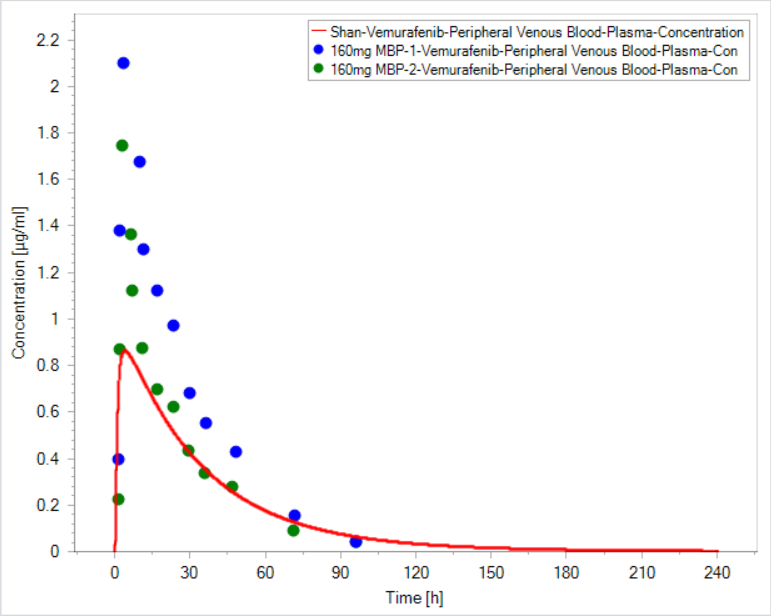
Hi, all
I am doing a vemurafenib (Log P 5.1, pka 7.1, MW 500, fu <0.01)simualtion, but I can't reach the Cmax like the observed data.
The hepatic clearance rate is according to publication value, and the fu is fitted to be 2.70E-4 to get consistent with blood plasma ratio of 0.72. The distribution model is Poulin & Theil. It's an oral formulation, however, choosing dissolved formulation can only make littele difference I have condiser transporters like ABCG2, ABCB1, OATP1B3 and also the EHC. After parameters identification, the parameters for transporters have been set to make the distribution in brain be of small fraction, which is agreement with publication, and biliary clearance get very small so that EHC might get insignificant.
Is there any else factor to better the fitting? Moreover, published data says vemurafenib is mainly excreted unchanged by biliary pathway, especially within first 48 h, and I think it suggest the EHC for vemurafenib is important. Can EHC influence the Cmax?
Any comment is appreciated.