 jackbibby1
commented
2 years ago
jackbibby1
commented
2 years ago Sorry, I'll also add that I've tried using: de.downProb = c(0, 1), and it seems like it's kind of working, but not really. e.g.
de_prob <- seq(0.1, 0.9, 0.1)
de_data <- list()
for (i in de_prob) {
cat("Generating data with", i, "de prob", "\n")
params <- setParams(sim_params, update = list("nGenes" = 200, "batchCells" = 1500,
"group.prob" = c(0.5, 0.5),
"de.prob" = i,
"de.downProb" = c(0, 1),
"de.facLoc" = 0.3))
sim <- splatSimulate(params, method = "groups")
df <- sim@assays@data$counts
df <- CreateSeuratObject(df) %>%
NormalizeData()
df$group <- sim$Group
de_data[[as.character(i)]] <- df
}
# I also just filtered out some of the de genes with small changes here, but it's broadly similar
sapply(mks, function(x) x %>% filter(p_val_adj < 0.05 & avg_log2FC > 0.4) %>% pull(cluster) %>% table()) %>%
t() %>%
data.frame() %>%
rownames_to_column("de") %>%
mutate(Group1 = -Group1) %>%
pivot_longer(cols = Group1:Group2, names_to = "direction", values_to = "genes") %>%
ggplot(aes(de, genes, group = direction)) +
geom_col(aes(fill = direction), col = "black", lwd = 0.3) +
labs(y = "Number of DE genes per group", x = "de prob") +
scale_fill_manual(values = c("tomato", "cornflowerblue"),
name = "Group") +
theme(panel.background = element_blank(),
panel.border = element_rect(fill = NA))

 lazappi
lazappi




Hi there,
Thanks for the great package.
I'm currently trying to simulate a bunch of expression files that contain 2 groups with increasing levels of differential expression. When I run this, it works well:
This is all good, showing increasing number of de genes with increasing de.prob value, and also that the number of de genes per group are pretty equivalent. I'm then trying to simulate similar datasets where the differentially expressed genes are unidirectional e.g. all de genes in the datasets are unregulated in Group1. To do this, I'm using the
de.downProbparameter (I hope I'm understanding the documentation OK?), setting it to either 0 or 1 e.g. :(using the same code as above to visualize the de genes)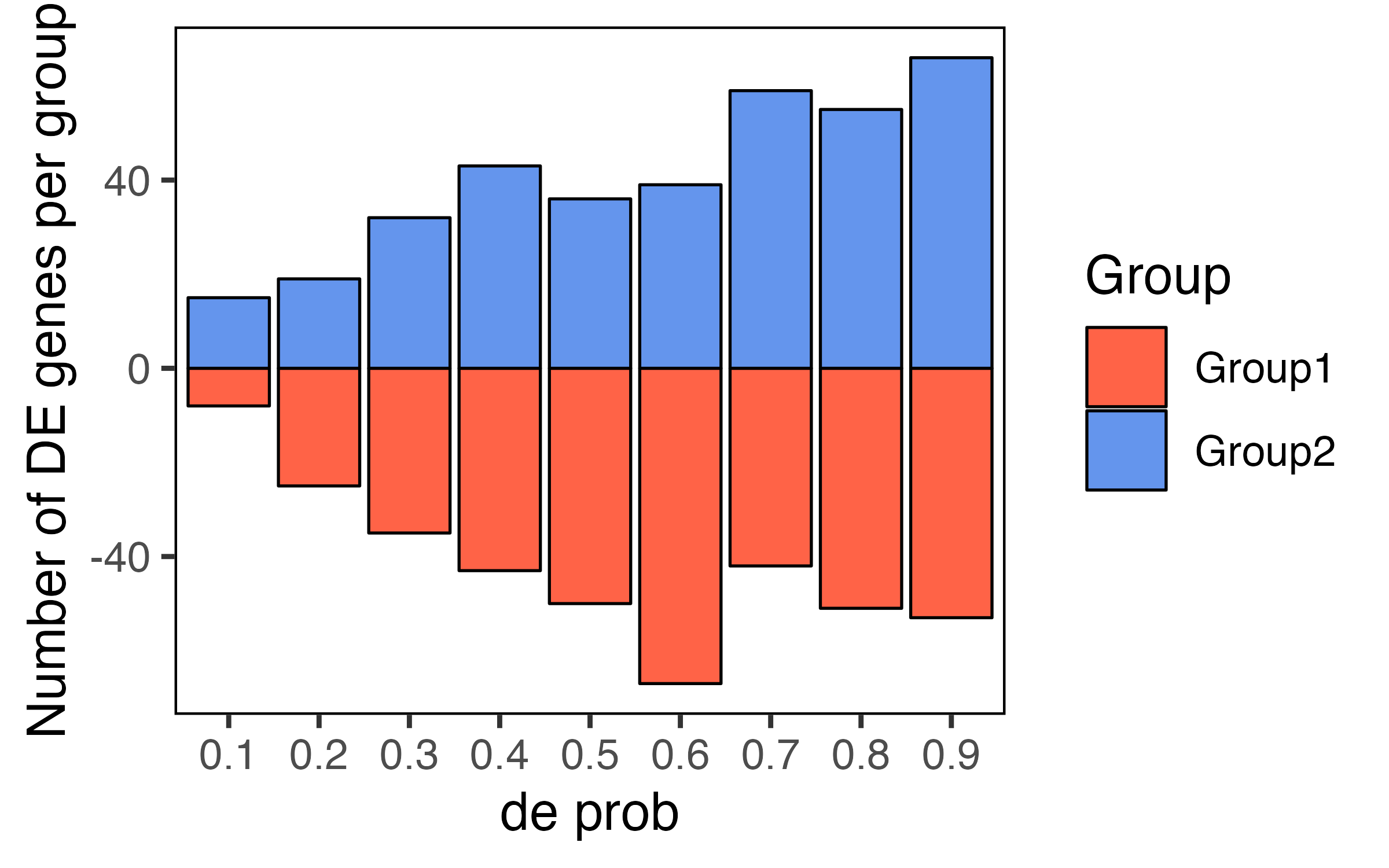
But this doesn't seem to work -- it ends up with a pretty similar distribution of directionality in de genes between the groups. Do you have an idea on what's going on here? Or can you suggest a better way to simulate the data where de genes are unidirectional?
Many thanks Jack