 PayamDiba
commented
2 years ago
PayamDiba
commented
2 years ago let's start with the clean simulated data (i.e. before adding technical noise). The clean data, since represents the true mRNA levels in each cell, does not requires library size normalization. So, you can try using the clean simulated data, as it is, for clustering. Also, it would be good if you could first run PCA on the clean data to extract the first few PCs (e.g. top 20 PCs) and then use that as an input for UMAP. Also can you color the cells according to their cell type assignment so that we could see the global structure. Please make these changes and let me know how the results look like. After that we can move to noisy simulated data.
 PeterZZQ
PeterZZQ The code that I used
The code that I used
 The code
The code
 But after I add the technical noise the result is like:
But after I add the technical noise the result is like:
 Here is the code for adding technical noise, should I adjust some parameters?
Here is the code for adding technical noise, should I adjust some parameters?
 anisioti
anisioti
Hi,
I wish to use sergio to benchmark the data imputation method. When I run the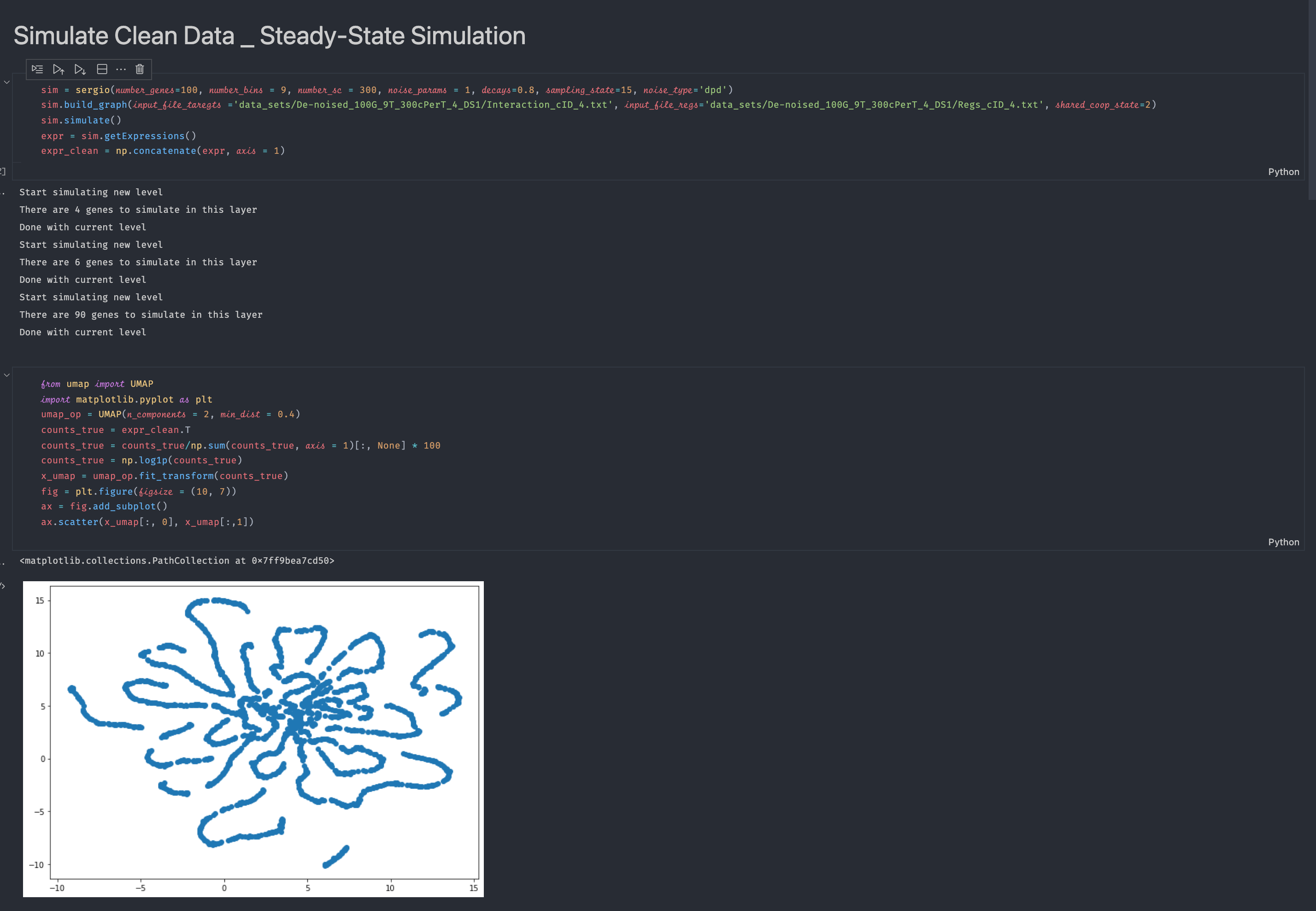
run_sergio.ipynbexample and visualize the clean expression data, I cannot see any clear cluster structure from the visualization (there should be 9 clusters according to the setting in the example function if I understand the readme file correctly). Please see the screenshot below for the code and visualization result:I also visualize the data after adding technical noise, still there is no cluster structure: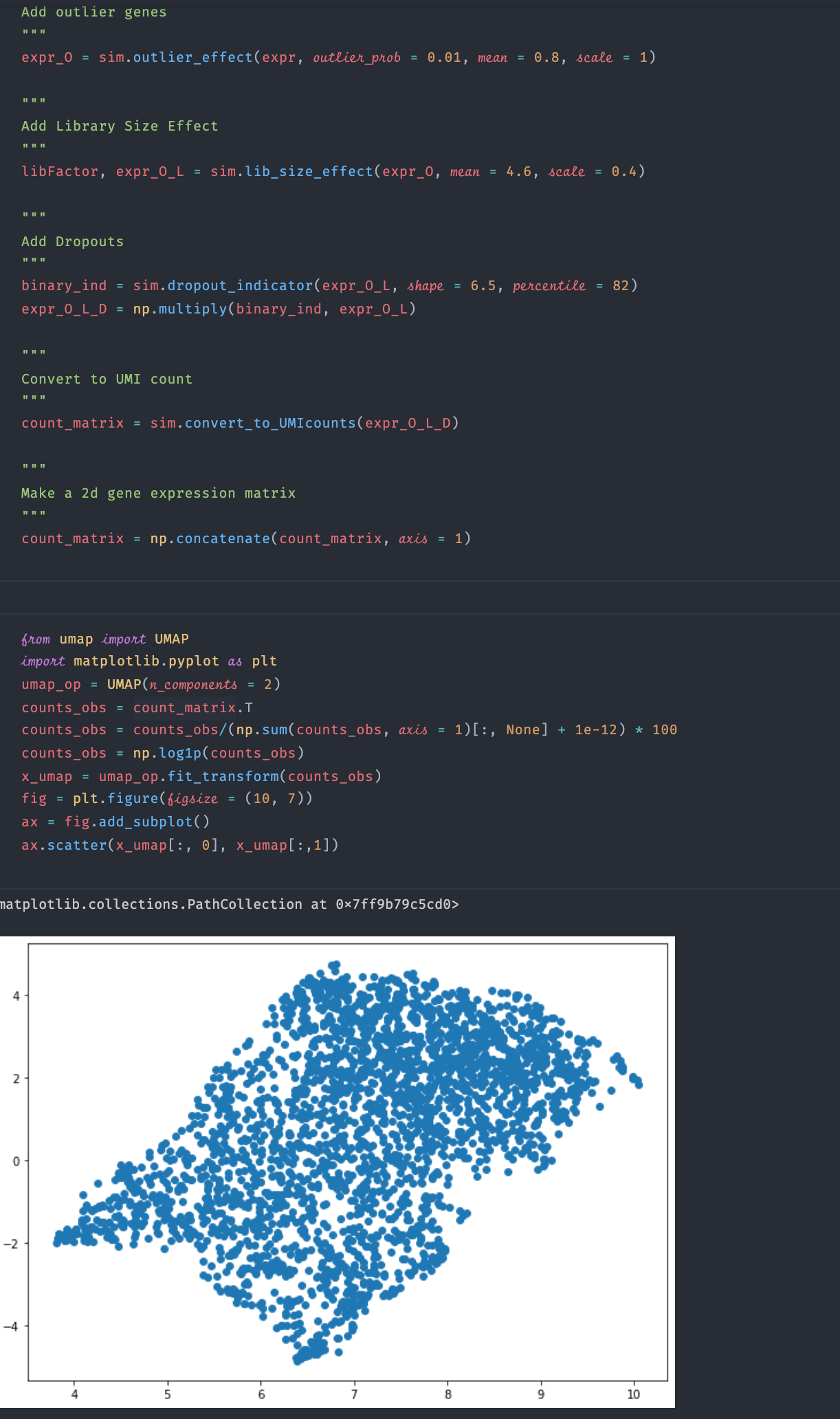
May I know how I can fix the problem? Thanks!