 yaaminiv
commented
5 years ago
yaaminiv
commented
5 years ago Created a choose-your-own-adventure-style slidedeck with information:
https://docs.google.com/presentation/d/1oE2c7QZGSeQ73HLsZw0hJBmLwODXwiVWfhYlJbzetMg/edit#slide=id.p
Open sr320 opened 5 years ago
yaaminiv
commented
5 years ago Created a choose-your-own-adventure-style slidedeck with information:
https://docs.google.com/presentation/d/1oE2c7QZGSeQ73HLsZw0hJBmLwODXwiVWfhYlJbzetMg/edit#slide=id.p
 kubu4
commented
5 years ago
kubu4
commented
5 years ago That's definitely thorough! Nice work!
Unfortunately, I think we wanted a lab-specific decision tree, so that we can use it for reference when deciding on things like:
Do we have enough DNA to do MBD?
What's the cost per sample on whole genome BS-seq vs. MBD BS-seq?
Which kit(s) do can we use for Illumina library construction, based on the amount of available DNA?
Basically, we need an easy way for us to figure out how we should perform BS-seq; enrich or not, make our own libraries or not.
yaaminiv
commented
5 years ago @kubu4
I think we wanted a lab-specific decision tree, so that we can use it for reference when deciding on things like:
- Do we have enough DNA to do MBD?
- What's the cost per sample on whole genome BS-seq vs. MBD BS-seq?
That information is already in the decision tree! I also included the MeDIP and MBD-Seq Qiagen kits we have.
- Which kit(s) do can we use for Illumina library construction, based on the amount of available DNA?
I'll add more information about that. Just so I know where to start, what kits have we used in the past?
yaaminiv
commented
5 years ago Added information about the Pico Methyl-Seq kit to the last slide but I can't find any other kits since Illumina's TruSeq kit was discontinued.
kubu4
commented
5 years ago I know it's in there but it's very difficult to find. In my opinion, something with fewer options would make the tree easier to use.
On Wed, Feb 13, 2019, 10:22 Yaamini Venkataraman <notifications@github.com wrote:
Added information about the Pico Methyl-Seq kit to the last slide but I can't find any other kits since Illumina's TruSeq kit was discontinued.
— You are receiving this because you were mentioned. Reply to this email directly, view it on GitHub https://github.com/RobertsLab/resources/issues/543#issuecomment-463309838, or mute the thread https://github.com/notifications/unsubscribe-auth/AEThOKEdTe2T59Obe3oHLdUcthcibx_Sks5vNFfMgaJpZM4aBvfN .
yaaminiv
commented
5 years ago I removed some options. Better?
 sr320
commented
5 years ago
sr320
commented
5 years ago This is certainly a great resource! At some point I would like to condense to a 1 pager - but suggest we do this in person. On Feb 13, 2019, 11:46 AM -0800, Yaamini Venkataraman notifications@github.com, wrote:
I removed some options. Better? — You are receiving this because you authored the thread. Reply to this email directly, view it on GitHub, or mute the thread.
kubu4
commented
5 years ago I'm going to add this to this issue, as it probably can be part of our decision tree. However, please look it over and see if my calculations make sense (and to see if I did this correctly!).
I performed a quick analysis to try to determine how much genome coverage we obtain from MBD sequencing (using @yaaminiv's C.virginica gonad MBD).
The TL;DR: 93% of genome is covered
Sequencing details:
Sequencing coverage
The process:
Input file: Bismark sorted BAM file
Generate coverage bedfile (via bedtools):
genomeCoverageBed -ibam cvir_bsseq_all_pe_R1_bismark_bt2_pe.sorted.bam -bga > bs_genome_cov.bed
In: head bs_genome_cov.bed
Out:
NC_035780.1 0 11 0
NC_035780.1 11 12 1
NC_035780.1 12 38 2
NC_035780.1 38 51 3
NC_035780.1 51 54 4
NC_035780.1 54 65 5
NC_035780.1 65 75 7
NC_035780.1 75 82 8
NC_035780.1 82 83 13
NC_035780.1 83 89 16Count total genome regions (i.e. lines):
In: wc -l < bs_genome_cov.bed
Out: 18779040
Count regions with no coverage (i.e number of lines where field 4 [coverage] equals zero:
In: awk '$4 == 0' bs_genome_cov.bed | wc -l
Out: 1481803
The math:
Percent of genome with no coverage:
Percent of genome with >0 coverage:
EDITED: Added sequencing deets
sr320
commented
5 years ago Curious - why not use .cov file Bismark produces?
kubu4
commented
5 years ago Don't know. Didn't consider looking at it - mostly because I hadn't looked into the format layout and knew that the bedtools had the builtin genomeCoverageBed tool that handles BAM files.
sr320
commented
5 years ago According C_virginica-3.0_CG-motif.bed there are 14458703 CGs total that are in play- (Total loci)
We want data on each CG.
(though maybe I am confusing this effort with the other issue - https://github.com/RobertsLab/resources/issues/609)
kubu4
commented
5 years ago Yes, I think you are confusing this effort with other issue.
My goal of doing the analysis above was to get an idea of what output differences in sequencing between WGBS vs. MBD, and then help guide us in deciding which sequencing approach to use.
On a side note, I looked at the documentation for the .cov files and it turns out the .cov files are 1-based coordinates. The BAM/BED files are 0-based. So, if the .cov files were used in @yaaminiv's analysis, that would be the easiest explanation for the off-by-one problem we saw.
sr320
commented
5 years ago I would still suggest we should be doing a comparison on CG loci basis as that what we ultimately examine. Yep you are correct on the 0-1 based shift. The main outstanding issue with her analysis is how the two strands are handled. She has levels for CG and GC on opposite strand which can and are often different and 1bp apart.
kubu4
commented
5 years ago Zymo input DNA requirements:
 jarcasariego
commented
5 years ago
jarcasariego
commented
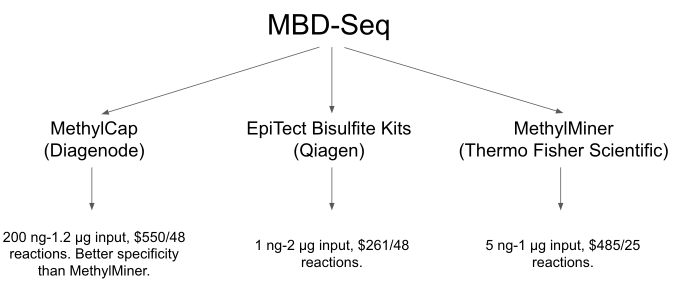
5 years ago Hi all, quick question. Why did you guys use the MethylMiner kit for MBD enrichment if you say in the flowchart that the MethylCap has a better specificity and the price is lower?
sr320
commented
5 years ago Hi all, quick question. Why did you guys use the MethylMiner kit for MBD enrichment if you say in the flowchart that the MethylCap has a better specificity and the price is lower?
Great question.. @yaaminiv would have to address as she would have included that comment re MethylCap - presume based on paper
@kubu4 any idea?
kubu4
commented
5 years ago Why did you guys use the MethylMiner kit for MBD
Most recently because (in no particular order):
jarcasariego
commented
5 years ago Then you had no experience with the MethylCap? Would you advise to give it a try? I'm very tight on budget and is more than half the price per sample of the MethylMiner.
kubu4
commented
5 years ago Correct, we have not used MethylCap. The initial format of the decision tree linked above ended up being a more comprehensive overview of available options for MethylSeq. We haven't yet pared it down to a lab-specific decision tree.
No reason not to try it.
The decision tree is still a work in progress and we're still assessing how much money (if any?) performing MBD saves us in sequencing costs.
jarcasariego
commented
5 years ago Got it. Thank you very much for your help. I'll stay tuned for updates on this great tool and sending some more questions for sure. Cheers...
Thinking about DNA quantity, targets, and cost I would like to develop a Diagram that outlines meth-seq options. Given @yaaminiv is in thick of this, could you please start this. Things we should consider include DNA quantity limits and cost estimates. Maybe Google Doc? @kubu4 can add his knowledge accordingly. Once a document is created drop a url in here and we can assist.