 kubu4
commented
5 years ago
kubu4
commented
5 years ago Thanks!
assemble MG7S2, but I can do this easily if you want it.
Yes please.
Closed emmats closed 5 years ago
kubu4
commented
5 years ago Thanks!
assemble MG7S2, but I can do this easily if you want it.
Yes please.
kubu4
commented
5 years ago Also, can you please post code used for all of your assembly/translation/etc?
kubu4
commented
5 years ago FYI, I've moved files to here, so that we can have URLs:
http://eagle.fish.washington.edu/oyster/index.php?dir=metagenomics_2019%2F
Please upload the remaining assembly to eagle/web/oyster/metagenome_2019 when you have it.
 emmats
commented
5 years ago
emmats
commented
5 years ago Sorry, put the last assembly in the original/wrong spot. The script for NGLess (assembly) is in the directory you indicated and is called ngl.singlelibs and was called by the command ngless.job. The single library assembly script is pretty much the exact same that I used for assembly of all the files.
The code for prodigal (translation) is: prodigal -i ../runNGL2/orf.fna -o out.prodigal -a proteins.prodigal.faa -p meta
kubu4
commented
5 years ago I've moved that file over to eagle/web/oyster/metagenome_2019.
These files look like they are just open reading frames. Do you have the raw assembly files, too? If yes, please upload them when you have a sec. Thanks!
emmats
commented
5 years ago all files are there in the metagenomics_2019 folder
kubu4
commented
5 years ago Do you have a singular assembly of all the samples combined?
emmats
commented
5 years ago Now in the metagenomics folder. contigs.fa
kubu4
commented
5 years ago Hey @emmats. I ran a comparison of your "co-assembly" and my "co-assembly" (i.e. all the samples combined into a single, large assembly). Here's the results:
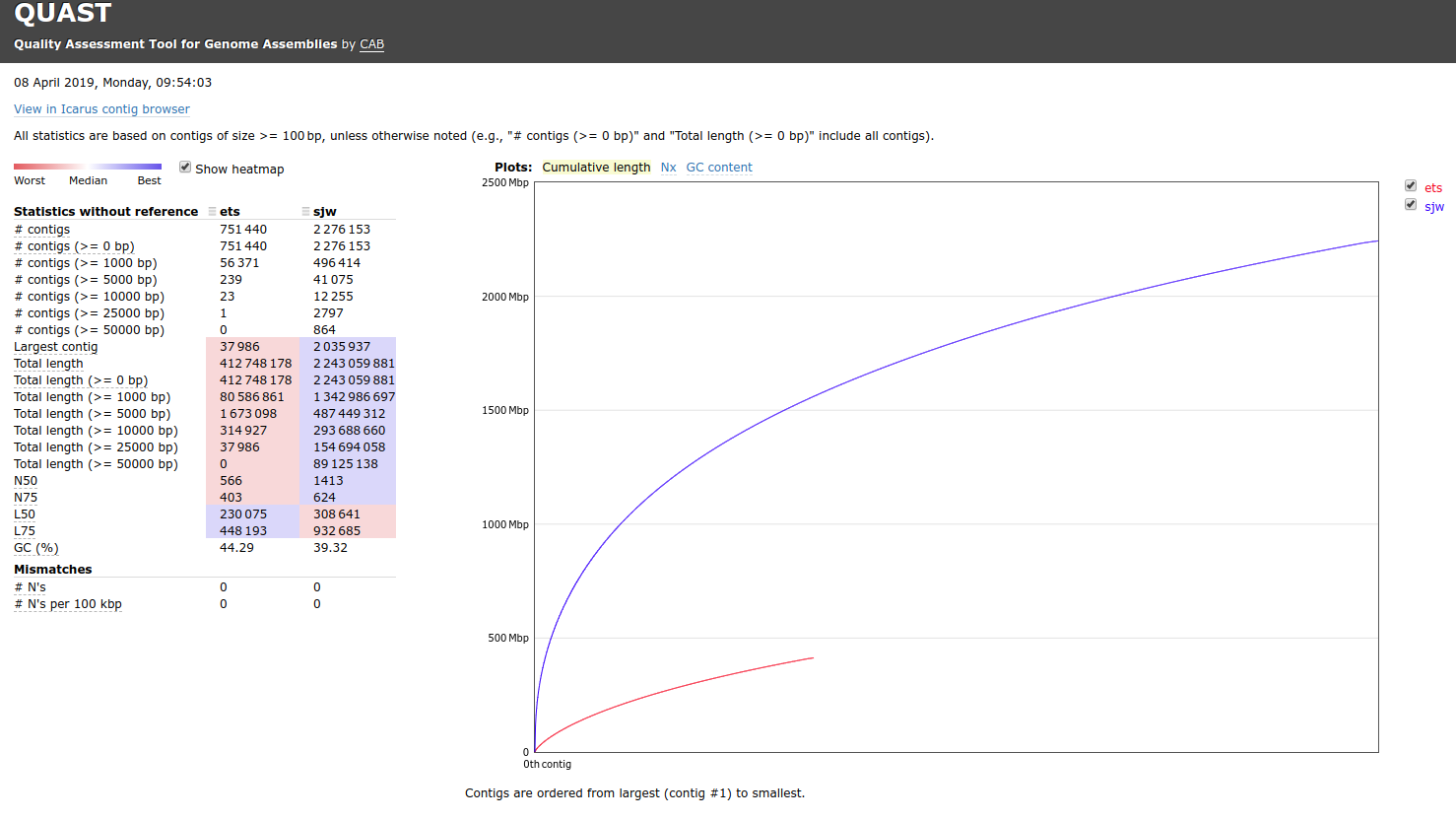
One thing that immediately jumps out at me is the "Total Length" (i.e. number of bases present in the assembly). My assembly is nearly 5x greater than yours. I feel like this can only be explained by:
Different input files
Different cutoff settings used in the assembler
I find it particularly surprising since I used Megahit to assemble these, and NGless also uses Megahit as its assembler!
Do you have the original script you used to generate this assembly so that we can double-check how it was made?
emmats
commented
5 years ago ngless "0.10" import "parallel" version "0.6"
samples = readlines('seq.files.list') sample = lock1(samples) input = fastq(sample)
output = preprocess(input, keep_singles=False) using |read|: read = substrim(read, min_quality=25) if len(read) < 45: discard
contigs = assemble(input) write(contigs, ofile='contigs.fa')
orfs = orf_find(contigs, is_metagenome=True) write(contigs, ofile='orf.fna')
kubu4
commented
5 years ago Do you have any thoughts on the differences seen in that data?
Also, can you please provide the contents of seq.files.list, as the script you posted above doesn't tell us which samples were processed.
emmats
commented
5 years ago I have no idea why they would be so different. Sounds like it is something worth sending to the developer? I can post it on their listserv, if you'd like.
cat seq.files.list Library_Geoduck_MG_1_S3_L002_R1_001.fastq Library_Geoduck_MG_1_S3_L002_R2_001.fastq Library_Geoduck_MG_2_S4_L002_R1_001.fastq Library_Geoduck_MG_2_S4_L002_R2_001.fastq Library_Geoduck_MG_3_S1_L002_R1_001.fastq Library_Geoduck_MG_3_S1_L002_R2_001.fastq Library_Geoduck_MG_5_S6_L002_R1_001.fastq Library_Geoduck_MG_5_S6_L002_R2_001.fastq Library_Geoduck_MG_6_S5_L002_R1_001.fastq Library_Geoduck_MG_6_S5_L002_R2_001.fastq Library_Geoduck_MG_7_S2_L002_R1_001.fastq Library_Geoduck_MG_7_S2_L002_R2_001.fastq
kubu4
commented
5 years ago Did you quality trim the FastQs for quality/adaptor removal?
emmats
commented
5 years ago No. Is that not included in the NGLess pipeline?
kubu4
commented
5 years ago I don't know. I didn't run NGless...
emmats
commented
5 years ago It says: preprocessing and quality control of FastQ files mapping to a reference genome (implemented through bwa by default) assembly of contigs annotation and summarization of the alignments using reference gene annotations
And from the paper: (1) preprocess the reads (performing quality-based trimming and filtering of short reads and, if appropriate, discarding reads that align to the host)
And I had commands for quality trimming in my script.
kubu4
commented
5 years ago Doesn't look like it handles adaptor removal. Not sure what the impacts of that might be. Maybe that's what we're seeing?
emmats
commented
5 years ago I'll see what I can find out from Luis.
emmats
commented
5 years ago Adaptor sequence trimming is not included. Although I'm not sure that would lead to the differences you saw.
Not as a builtin function, however since several good tools exist (trimmomatic, cutadapt, SeqPurge, etc.) the most flexible option would be to create external modules to allow calling them from ngless. There's a repository of external modules contributed by the community at https://github.com/ngless-toolkit/ngless-contrib/ .
I've also created the issue: https://github.com/ngless-toolkit/ngless-contrib/issues/1 to track this. I might get around to add this external module but regardless contributions are welcome.
Hope this helps, Renato
I put the files on eagle under the emma folder. Here is what you have: orf.fna - entire metagnome assembly proteins.prodigal.faa - translation of metagenome assembly orfMG...fna - assemblies of individual metagenomes (individual tanks & days). For some reason I didn't assemble MG7S2, but I can do this easily if you want it.