 zasdfgbnm
commented
5 years ago
zasdfgbnm
commented
5 years ago Hi @maxentile, I see this issue and thank you so much for this very detailed explanation. Your feedback is important to us. I will try to reproduce and get back to you as soon as possible.


 maxentile
maxentile
 isayev
isayev jchodera
jchodera Jussmith01
Jussmith01






Hello,
Thank you very much for making this project available!
We are excited about the prospect of using ANI-1ccx for tautomer ratio prediction, but have started encountering some surprising behavior when designing and testing Monte Carlo moves for this purpose.
Tagging Marcus Wieder (@wiederm) who helped prepare this Issue with me, and John Chodera (@jchodera) who may be interested in following this thread.
1. Existence and favorability of unphysical local minima on methane example
1.1. Bond-length scan from a tetrahedral geometry
We started by inspecting the methane example
If we take an ideal tetrahedral geometry and scale all the C-H bond lengths, we obtain the following energy profile:
The minimum-energy tetrahedral geometry of methane along this scan is approximately:
with a C-H bond length of 1.107 angstroms, and an energy of -106266.1875 kJ/mol.
1.2. Accessibility of unphysical local minima in room-temperature MD
We ran 50 picoseconds of constant-temperature MD at 300K starting from the tetrahedral geometry obtained above.
(Code: https://gist.github.com/maxentile/ac04d1fab8dd4aa217fe224190d06765)
The simulation quickly finds many geometries with lower apparent energy than the starting tetrahedral geometry.
The simulation does not retain a tetrahedral geometry. Instead the H-C-H angles appear to be metastable.
Here are the histograms of H-C-H angles visited in the trajectory:
Also the C-H distances appear to be bistable: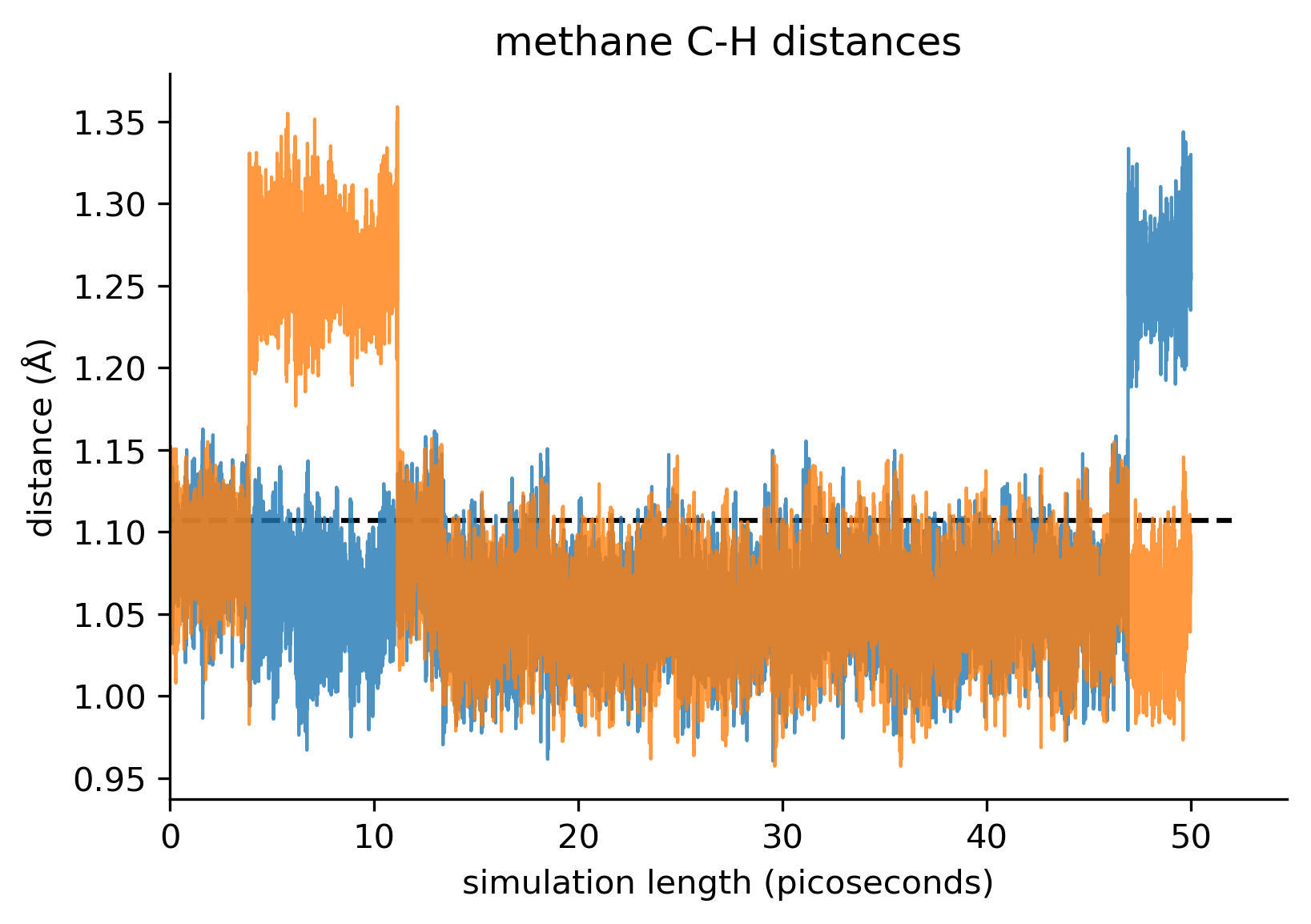
1.3. ANI-1ccx predicts a non-tetrahedral geometry to have nearly 50 kJ/mol lower energy than the minimum-energy tetrahedral geometry
The lowest-energy snapshot from this trajectory includes nearly right angles, and is over 40 kJ/mol more favorable than the minimum-energy tetrahedral geometry. We further minimized its energy using gradient descent on the ANI-1ccx potential, obtaining the following:
with two of the C-H bonds at length 1.27 angstroms and the other two C-H bonds at length 1.07 angstroms.
The H-C-H angles at this conformation are 92.2 , 102.5, and 143.5 degrees.
The ANI-1ccx energy of this configuration is -106313.8125 kJ/mol.
This is ~47 kJ/mol more favorable than the minimum-energy tetrahedral geometry.
Is there an intuition for this behavior, and is anything similar observed for more complicated molecules?
1.4. Energy scans
We took the energy-minimized geometry and scaled it up and down, obtaining the following energy profile (in orange):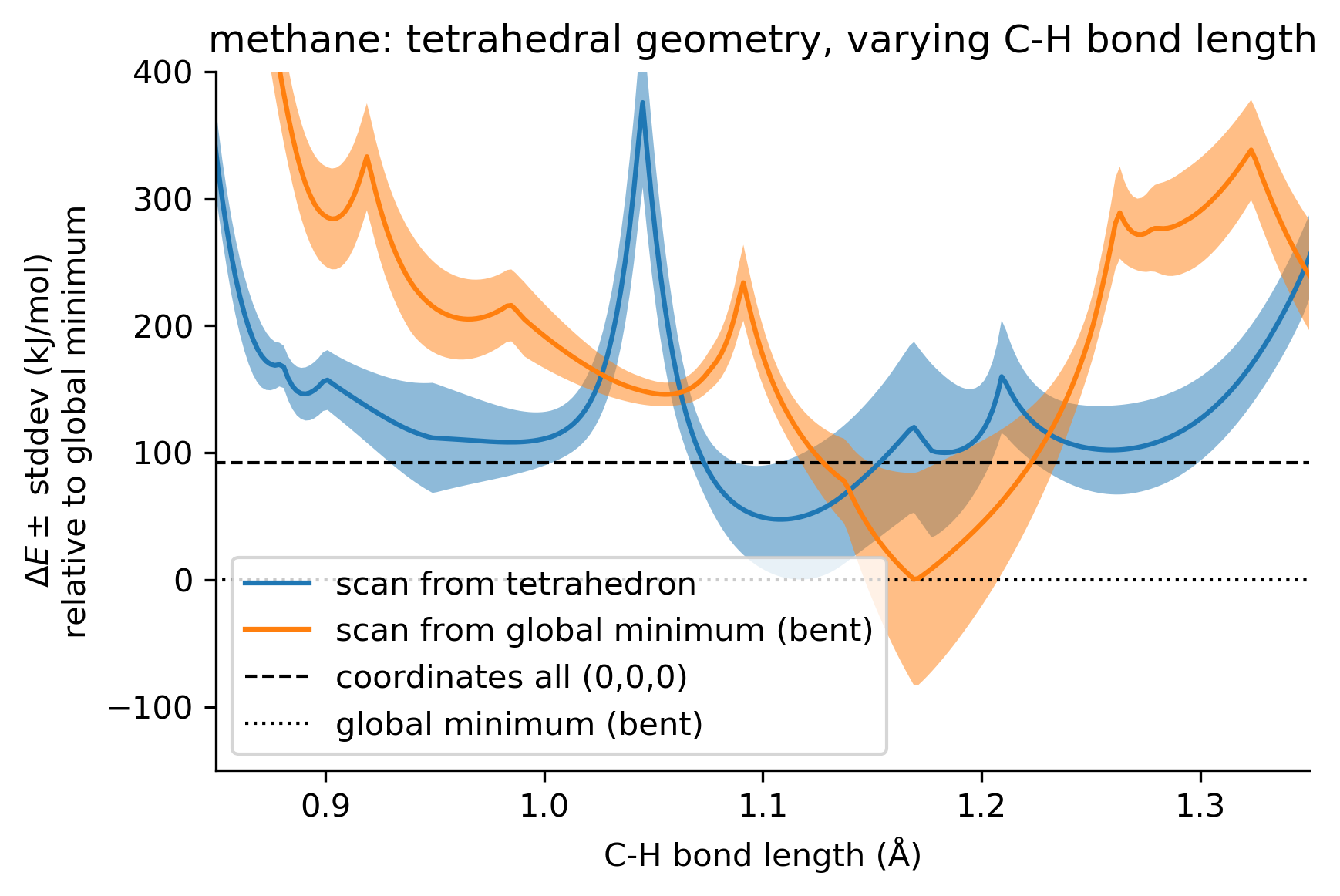
The x-axis represents the average C-H bond length. The uncertainty bands represent the standard deviation among the 8 models in the ANI-1ccx ensemble.
For comparison, in blue is the energy profile we obtained for tetrahedral geometries, the dashed line is the energy obtained when the coordinates are all (0,0,0), and the dotted line at 0 corresponds to the global minimum energy encountered so far.
We observe that the energy profiles have many local minima and cusps. Is there an intuition for why the energy profile looks this way, or how it might be corrected?
Also, it appears that the ANI-1ccx ensemble has large uncertainty throughout these scans -- is there guidance on how to use this information?
2. Local energy minimum when all coordinates are zero
We further noticed that for the methane example in the
Computing Energy and Forcetutorial https://aiqm.github.io/torchani/examples/energy_force.html, setting all coordinates to (0,0,0) is ~30kJ/mol more favorable than the the starting geometry (-106221.367 kJ/mol vs -106189.195 kJ/mol).2.1. Energy scan, scaling all C-H distances
For illustration, here is the energy profile as we scale the example starting geometry up and down: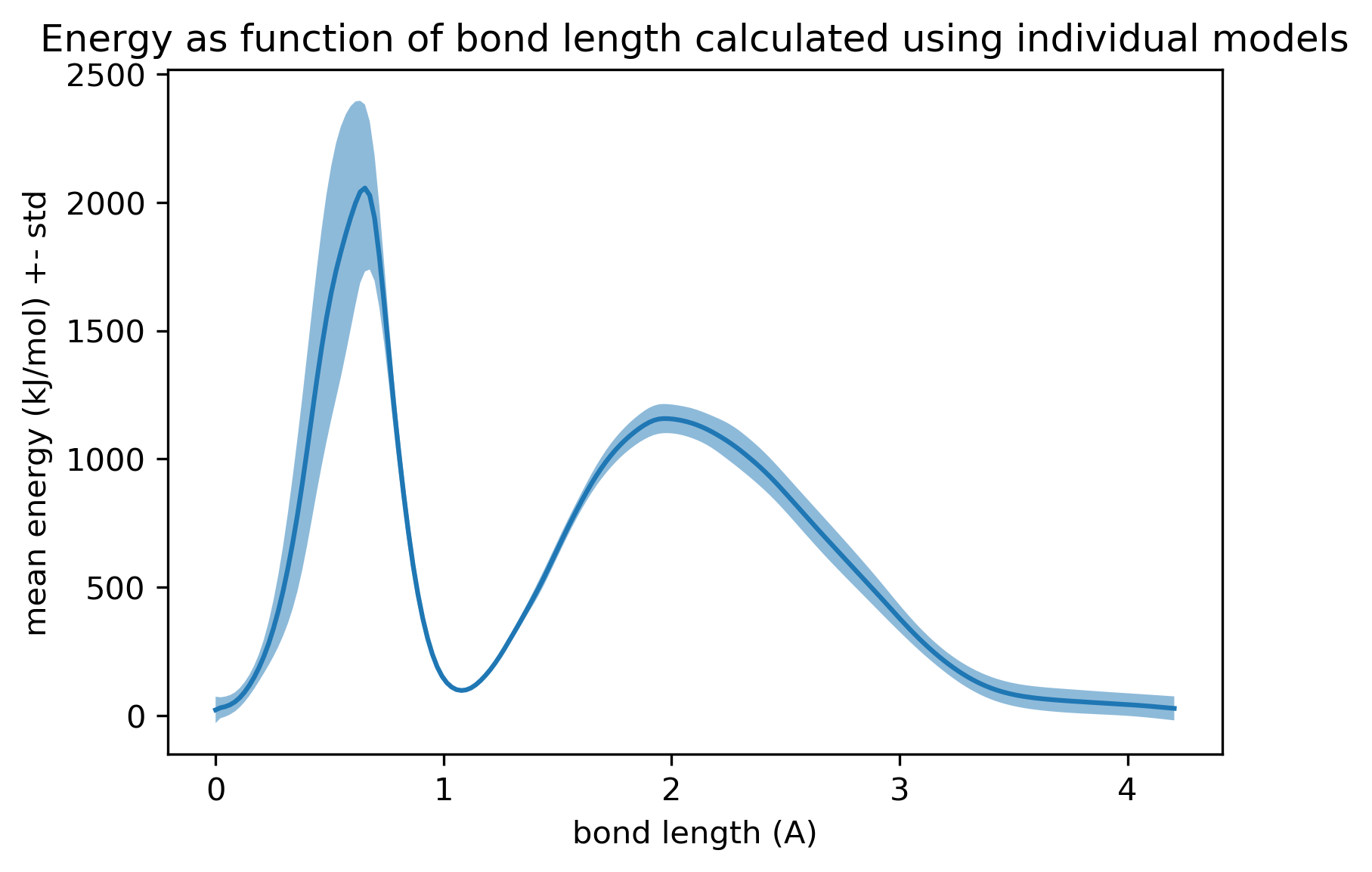
The existence of a local minimum with all nuclei on top of each other is concerning in the context of Monte Carlo moves that instantaneously propose to move hydrogens, since they might randomly propose geometries that induce steric clashes, and we would want such proposals to be rejected.
2.2. Comparison: changing only one H position, rather than all H positions
If we bring only one of the hydrogens closer to the central carbon (rather than all of them), we obtain an energy profile that instead looks like this: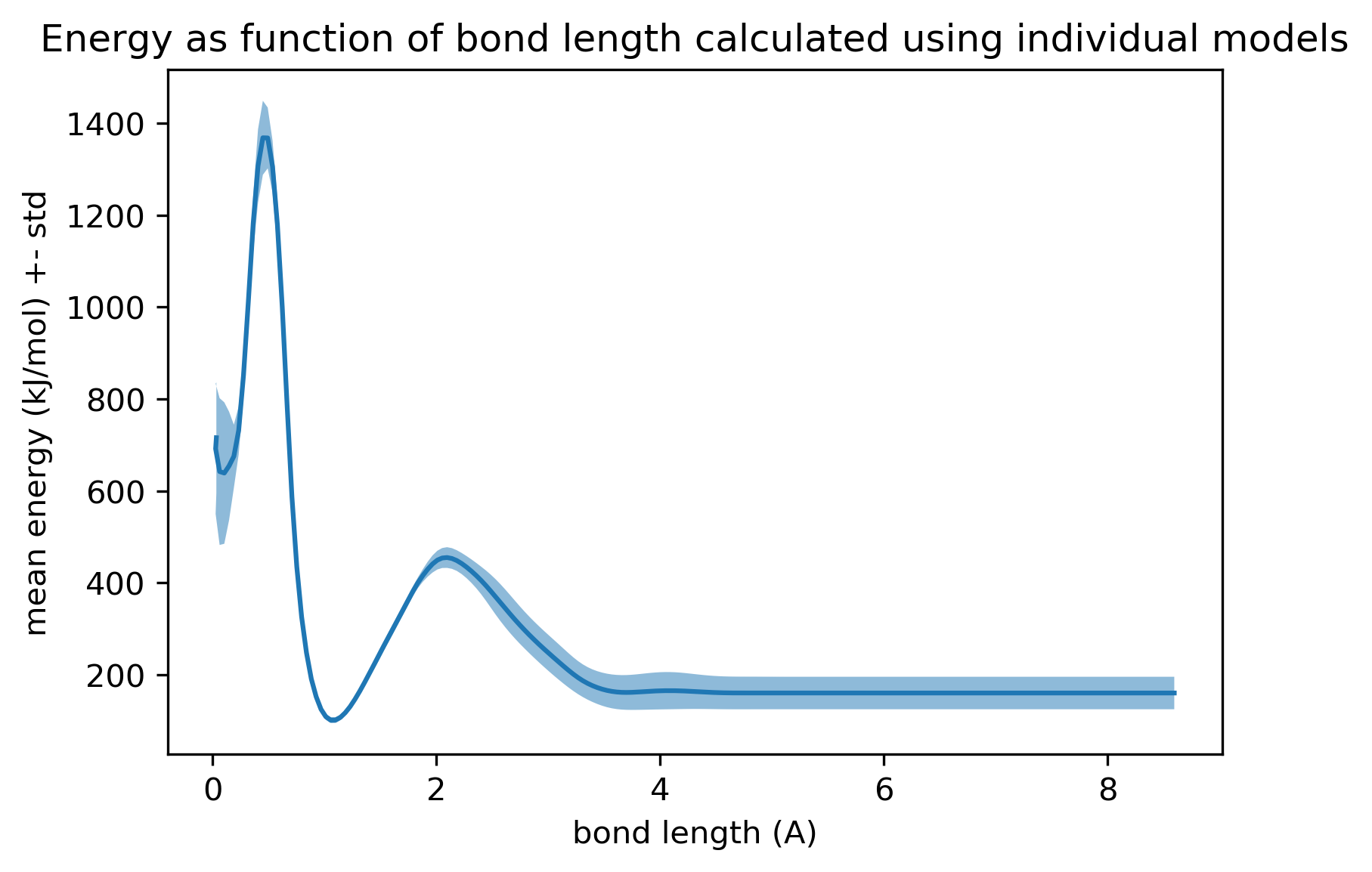
The high energy predicted when one hydrogen is placed on top of the central carbon is reassuring, since it suggests instantaneous Monte Carlo proposals that try to place one hydrogen directly on top of a heavy atom will not be accepted with any appreciable probability. However, it is still surprising that the energy does not increase monotonically as the interatomic distance approaches zero from the equilibrium bond length.
Is there an intuition for how to correct this behavior?
Best regards, Josh