 apcamargo
commented
9 months ago
apcamargo
commented
9 months ago Hi @elozanoe
Score calibration is blocked by default for samples with less than 1,000 sequences because the calibration works well when you have lots of sequences (see this figure) because the estimate of the underlying composition is more reliable.
How many genomes you have? My recommendation is to concatenate all the assemblies into a single FASTA to have as many contigs as possible in a single input. You can then use the --force-auto parameter to force score calibration regardless of the number of sequences. I've never benchmarked calibration with few sequences extensively, but my guess (based on the figure I linked above) is that it would still improve classification performance as long as you have at least 400-500 contigs. It might work well enought with even less than that, but I'd take a careful look at the results just to be sure.
Please let me know what you find out!
 elozanoe
elozanoe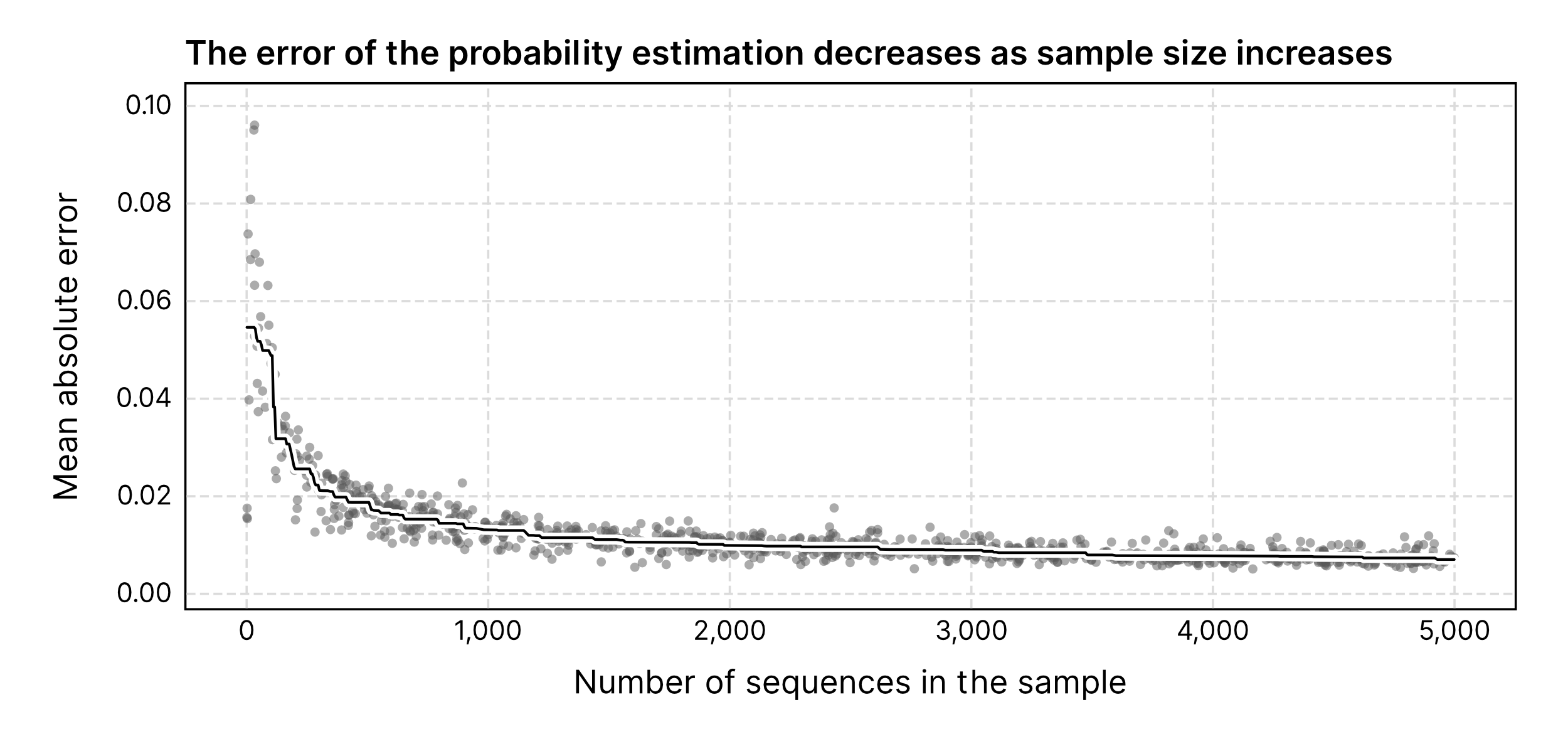{kind=link}
Hello. I've been reading the documentation and the use of --enable-score-calibration is not clear to me. When I apply it in the end-to-end pipeline, it tells me that I have less than 1000 sequences and that another option will be used by default if I do not apply an automatic option.
I am working with prokaryotic genome assemblies (less than 1000 contigs) and I would like to know the estimated probabilities. What is the most recommended option? Should I do genomad end-to-end without any tags?
Thanks in advanced!