 benjjneb
commented
7 years ago
benjjneb
commented
7 years ago There is a solution for this problem in the dada2 package: collapseNoMismatch performs "100%" OTU clustering, i.e. collapses all sequences that differ only be length differences. However, the current implementation is much slower than necessary and should be improved (I hadn't seen anyone bring up a natural use case yet!).
In general we recommend cleaning up the artificial variation before applying dada(...) for best results though. With 3% OTUs you could often "get away" with leaving artificial variation in the data, as it would hopefully get lumped into the same OTU as the biological sequence. But with dada2 (or any other method that infers ASVs exactly) artificial variation, such unremoved primers with ambiguous nucleotides or untrimmed heterogeneity spacers, will often look like biological variation to the algorithm.
 joey711
joey711 werdnus
werdnus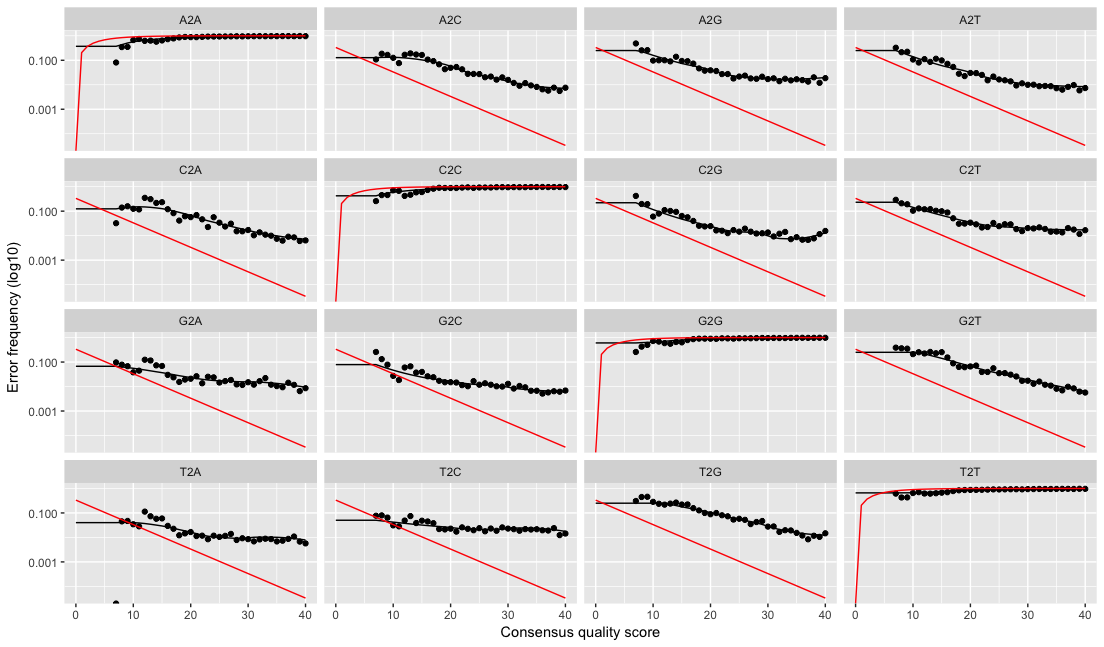
 FabianRoger
FabianRoger
I've been helping a friend work through using DADA2 with a project that was sequenced across two different MiSeq runs. An interesting aspect of his project is that nearly all samples from the first run were re-sequenced in the second run, so it gives us a great opportunity to examine the performance of various sequence processing pipelines in merging runs — you would expect the same vial of DNA to yield roughly similar sets of amplicon sequences when resequenced.
Working with a subset of samples, we found that a QIIME workflow with uclust gave us exactly what we expected: the OTUs picked individually had the same refseqs, and when picking OTUs with both runs at the same time, reads were split about evenly between the runs for each OTU (though the second run had greater sequencing depth, so there's a tail of rare OTUs present only in that run). Switching to DADA2 with the same sample was distressing: we found that all the variation collapsed, and
dada()infers just one sample sequence from the ~5000 unique input sequences. Well, given that this is a seed endophyte study, I was prepared to believe that most of the variation was error and the community was dominated by just one taxon, but the problem was that the inferred sample sequence was different for the two different runs!I thought that perhaps including some more samples for learning error rates might fix the issue. Instead, I was able to infer more sample sequences, but they were still segregating by run, even though I knew from the uclust results that the sequences were the same.
Pulling the two inferred sequences from the single-sample trial, I was distressed to find that they were exactly the same, save for one sequence being just a single base-pair longer on the 5' end.
That's odd, particularly since these sequences have already been trimmed (by the sequencing facility) to remove the adaptors and sequencing primers. So, I pulled a bunch of sequences out of the sample and had a look at how they aligned:
It turns out that there's a lot of variation in where the sequence starts and ends — the alignment has "raggedy" ends. I talked to my friend about this, and he mentioned that this was intentional: something the sequencing facility did to introduce heterogeneity into the sequencer. They place a 1–10 bp variable spacer after the sequencing primer, randomly, to keep from swamping the sequencer with all the same base at the same time. They're suppose to be removed with the sequencing primer!
Well, clearly they're not, and they're introducing small artificial differences at the ends, which is a problem (though easily fixable by trimming ~10bp from each end), but they're also keeping DADA2 from recognizing that it's the same RSV in both runs, which is a much bigger problem.
I solved the problem by including the
pool=TRUEoption when performing inference:I think the way to solve the issue more elegantly will be to use the stable region at the beginning and ends of the amplicons (these are V5-V6-V7 16S sequences) to trim (pretending that they're primers), so that all of the sequences have the same start and end point. Pooling for sample inference across the full set of samples is a little bit computationally intensive, to say the least. It'll require the cluster, if that's the route my friend takes with this.
I'm putting this up because it was an issue I struggled with for days, and I wanted to put it out to the community. If you're having trouble merging sequencing runs with DADA2, check the alignment of your sequence ends: I'm not sure how prevalent this inclusion of variable length heterogeneity spacers in the primer thing is, but it seems to be gaining popularity.