 GeorgescuC
commented
2 years ago
GeorgescuC
commented
2 years ago Hi @andynkili ,
The infercnv analysis forks in two branches after a certain point as illustrated on the wiki. On one side, the final plot shows the residual expression that you can also see after step 15 (preliminary plot) once it has been denoised. This plot should always be the first you look at to check for any issues in the levels of noise remaining in your data, to verify that your references are properly clustered and don't exhibit signal themselves, and that the (sub)clustering of your tumor cells makes sense once you look at the HMM. On the other side, the HMM plots from step 17-20 are on a different "track" of the analysis where the spiked-in data is used to calibrate the HMM to predict CNVs from the residual expression (before denoising). Overall the predictions seen at step 17 look in accordance with what you can estimate from looking at the residual expression so there does not seem to be any issue. Once the Bayesian filtering is done however, you are left with very few CNVs, which I would expect to be the combined result of not using references and a low threshold for the filtering. This filtering is based on the Posterior probabilities, not pvalues.
Also, for the chromosome bars issue, there is now a fix available on the master branch. The issue stems from the input gene coordinates file having contigs/chromosomes that have no genes on them (initially or post filtering) that "eat" the unique colors defined for the actual chromosomes. These contigs still exist as valid levels in the factor list in R, but are not counted when checking for unique(contigs).
You can either:
- update your install of infercnv and rerun your dataset.
- filter your input annotations with a variant of this command
grep -v -P "[GJ][LH][0-9]+[_random]?" ncbi_mm10.txt > ncbi_mm10_filtered.txtthen rerun infercnv. - manually fix the levels in the infercnv object with
infercnv_obj@gene_order[["chr"]] = droplevels(infercnv_obj@gene_order[["chr"]]).
Regards, Christophe.
 andynkili
andynkili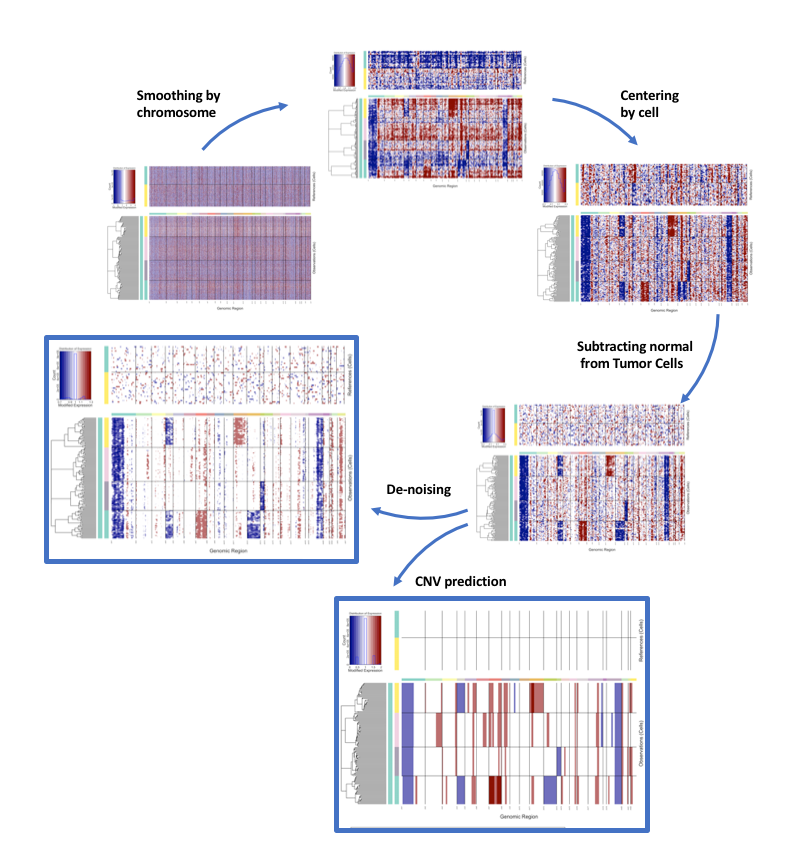{kind=link}
Hi, First, thank you again for this wonderful tool. Second, I don't understand why some CNVs predicted at step 17 and removed after step 19 are still present in the final plot:



How come plots from step 19 and 20 only shows mainly CNVs on chromosome 19, but in the end the final plot has a lot more CNVs (on Chr1 or Chr5 for example)? Aren't those supposed to be classified as normal states by the bayesian mixture model?
Best, Andy