cwalker7
commented
4 years ago
cwalker7
commented
4 years ago My code for running the simulations is something like this:
`import numpy as np
from simtk import unit
from openmmtools.multistate import MultiStateReporter, MultiStateSampler, ReplicaExchangeSampler
from openmmtools.multistate import ReplicaExchangeAnalyzer
total_simulation_time=20.0*unit.nanosecond
simulation_time_step=10*unit.femtosecond
exchange_frequency=100
simulation_steps = int(np.floor(total_simulation_time / simulation_time_step))
exchange_attempts = int(np.floor(simulation_steps / exchange_frequency))
num_replicas = len(temperature_list)
sampler_states = list()
thermodynamic_states = list()
for temperature in temperature_list:
thermodynamic_state = openmmtools.states.ThermodynamicState(
system=system, temperature=temperature
)
thermodynamic_states.append(thermodynamic_state)
sampler_states.append(
openmmtools.states.SamplerState(positions)
)
move = openmmtools.mcmc.LangevinDynamicsMove(
timestep=simulation_time_step,
collision_rate=1.0/unit.picosecond,
n_steps=exchange_frequency,
reassign_velocities=False,
)
simulation = ReplicaExchangeSampler(mcmc_moves=move, number_of_iterations=exchange_attempts)
reporter = MultiStateReporter("output.nc", checkpoint_interval=1)
simulation.create(thermodynamic_states, sampler_states, reporter)
simulation.minimize()
simulation.run()
analyzer = ReplicaExchangeAnalyzer(reporter)
(
replica_energies,
unsampled_state_energies,
neighborhoods,
replica_state_indices,
) = analyzer.read_energies()
# Convert replica_energies to state_energies
n_particles = np.shape(reporter.read_sampler_states(iteration=0)[0].positions)[0]
T_unit = temperature_list[0].unit
temps = np.array([temp.value_in_unit(T_unit) for temp in temperature_list])
beta_k = 1 / (kB.value_in_unit(unit.kilojoule_per_mole/T_unit) * temps)
n_replicas = len(temperature_list)
for k in range(n_replicas):
replica_energies[:, k, :] *= beta_k[k] ** (-1)
total_steps = len(replica_energies[0][0])
state_energies = np.zeros([n_replicas, total_steps])
for step in range(total_steps):
for state in range(n_replicas):
state_energies[state, step] = replica_energies[
np.where(replica_state_indices[:, step] == state)[0], 0, step
]
# Physical validation is run on 'state_energies' for adjacent temperature pairs` mrshirts
mrshirts jchodera
jchodera jaimergp
jaimergp

 Lnaden
Lnaden It is also about 13% faster than ReplicaExchangeSampler this case.
It is also about 13% faster than ReplicaExchangeSampler this case.
 asilveirastx
asilveirastx
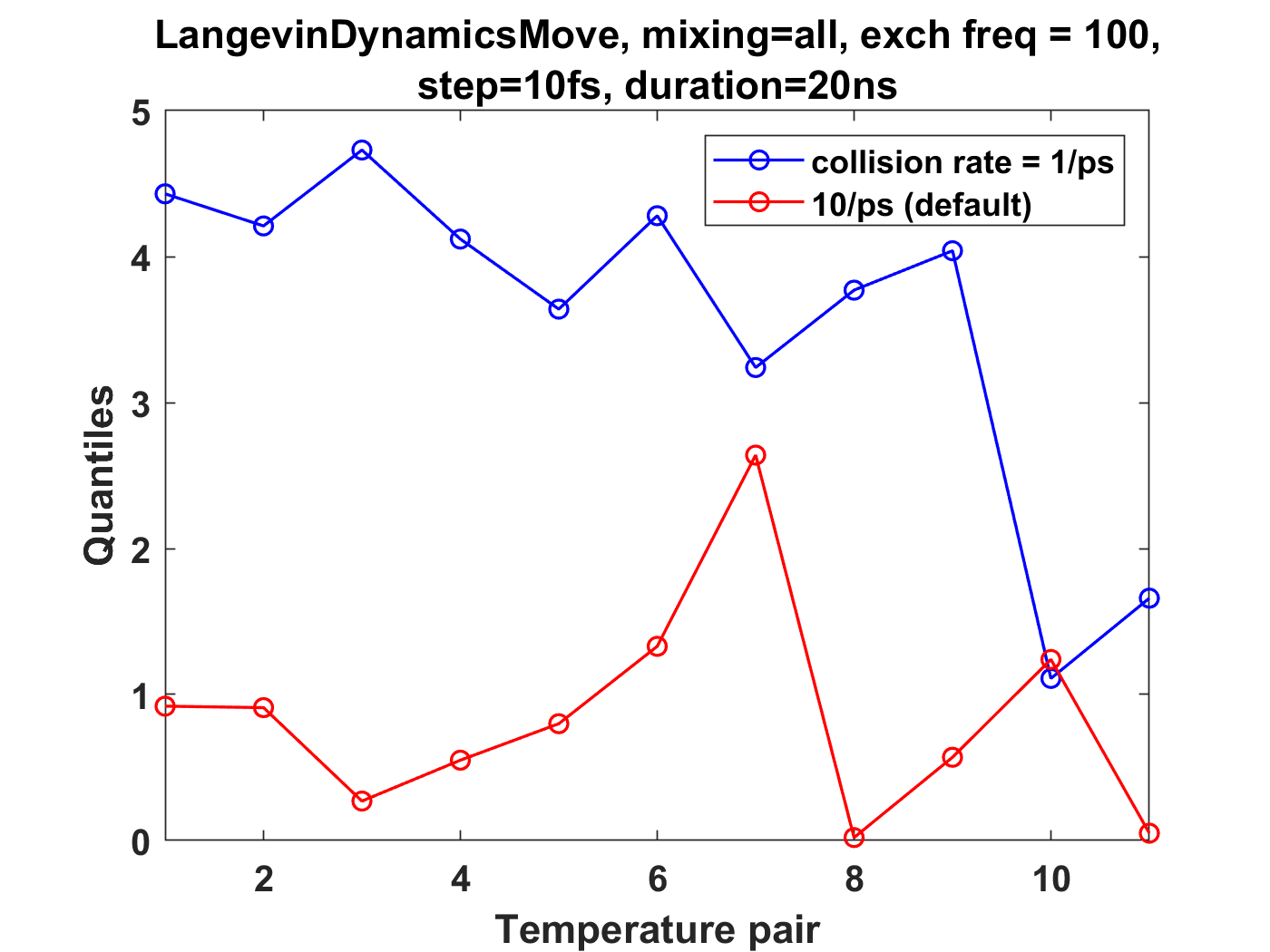
I am running temperature replica exchange simulations on small coarse-grained oligomer systems (containing a single chain), and have been using an ensemble validation check on the energies for thermodynamic state pairs. This is from the physical-validation python package (https://physical-validation.readthedocs.io/en/latest/userguide.html#ensemble-validation). I am finding that for a system with 12 replicas spaced logarithmically from 200K to 300K, using LangevinDynamicsMove with replica_mixing_scheme='swap-all', n_steps=100, collision frequency=1/ps, and time_step=10fs, the ratio ln(P(U)_1/P(U)_2) is consistently about 4-5 standard deviations off from the analytical value. The physical validation code uses the pymbar DetectEquilibration function to trim the energies for each state.
I don't think it's an issue of the time step being too large, as reducing to 1fs (with n_steps set to 1000) still leads to 4-5 standard deviations for the above system.
I've tried a number of different replica exchange parameter variations: Upon increasing n_steps to 1000, increasing the friction to 10/ps, or setting the mixing scheme to 'swap-neighbor', there is much better agreement (~1 standard deviation or less from analytical value). Those are comparable to the results for replica_mixing_scheme=None (no exchanges), and for independent simulations run in openmm.
My question is - is this a matter of simply not equilibrating each replica long enough after an exchange, or could there be an underlying issue somewhere with the exchanges?
These plots summarize the results, where quantiles is standard deviation from the analytical ratio: