 FedeGueli
commented
2 years ago
FedeGueli
commented
2 years ago Gisaid Queries: Spike_N460K,Spike_G476S,M_Y71H or M_Y71H,N_T135I find all the three sequences.
Closed TakaKen6 closed 2 years ago
FedeGueli
commented
2 years ago Gisaid Queries: Spike_N460K,Spike_G476S,M_Y71H or M_Y71H,N_T135I find all the three sequences.
FedeGueli
commented
2 years ago 14853710 has S:K356T while 14853841 has S:G257V
 agamedilab
commented
2 years ago
agamedilab
commented
2 years ago  corneliusroemer
commented
2 years ago
corneliusroemer
commented
2 years ago Thanks @TakaKen6 for opening this issue and @FedeGueli @agamedilab for providing additional insights.
These are the mutations on top of the BA.2 founder:

BA.2.3.2 is defined by C26408T = E:S55F, so Usher is probably wrong here as S:460K is homplasic, where E:S55F is less likely to be so. This is likely a true BA.2.3.2
Mutations on the branch from BA.2.3.2 are: T19644C, C27752T, C151T, G293A, C629T, A2442C, C4763T, A5405G, A5648C, C6573T, C9979T, A10393C, C11750T, C15654T, C16418T, C20946T, G22599C, A22629C, T22917G, T22942G, G22988A, G23040A, C23481T, T26733C, C27707T, C28677T, nuc 21992/21994del, 27266-27300/del
ORF1a E10K, L122F, E726A, H1500Y, I1714V, K1795Q, S2103F, L3829F
ORF1b T984I
S R346T, K356T, L452R, N460K, G476S, R493Q, S640F
S gaps Y144-
S insertion 212:NGE (thanks @FedeGueli for noticing)
M Y71H
ORF6 gaps 22/33- -> frameshift that has been observed before, likely viable
ORF7a A105V, T120I
N T135I
The Spike profile is convergent with what we've seen happen in BA.5/BA.2.75 and other growing lineages so this is definitely one to keep an eye on. In particular having 346T, 356T, 460K and the 493Q reversion makes this concerning from an immune escape perspective.
Interestingly there are many ORF1a mutations as well - ORF1a:K1795Q was common in Gamma.
I'd say we designate as soon as we see it pop up again, ideally in another country.
FedeGueli
commented
2 years ago @corneliusroemer E:S55F is homoplasic too, especially in BA.2 popped up several times but also in BA.5 , i think @alurqu proposed and then closed something with that cause wasnt monophyletic. As you say likely it is BA.2.3.2 but i will not be 100% sure Usher is wrong.
corneliusroemer
commented
2 years ago Fair point @FedeGueli - but BA.2.3.2 was at ~40% in Vietnam which is where the travellers came from so on the balance of probability it's much much more likely Usher pulled it onto the more homoplasic S:460K (we know this is very homoplasic in current second gens).
 FedeGueli
commented
2 years ago
FedeGueli
commented
2 years ago @corneliusroemer agreee with you, even if there is some back rumour in my mind on the fact S:460K could permit more mutations to be accumulated so it would make sense that a hypothetical BA.2.3 + S:460K ancestors gave birth to these' heavily' mutated variants (referrimg to BA.2.3.20 ), but at this time pure speculations. So scratch that and agree going on with BA.2.3.2 is the more reasonable placement.
 alurqu
commented
2 years ago
alurqu
commented
2 years ago @FedeGueli You are referencing Issue 760 for BA.2.12.1+E:S55F. And yes I closed it because the UShER tree was far from monophyletic.
I'll also note that I originally thought BA.2.3.17 was a BA.2.3.2 due to the E:S55F mutation. The discussion in the BA.2.3.17 ticket might be helpful.
I did look, and these sequences do not have BA.2.3.17's ORF1a or ORF1b mutations.
 cvejris
commented
2 years ago
cvejris
commented
2 years ago This lineage is indeed (a little bit remotely) sister to BA.2.3.20. What constitutes the background mutational constellation from which these two saltatory lineages emerged is precisely the couple 460K and 493Q, which are both also found in BA.2.75 - what a coincidence :)

 c19850727
commented
2 years ago
c19850727
commented
2 years ago @corneliusroemer I personally regard ORF1a:K1795Q as a marker of chronic infection, and it's associated with a few genomes that have convergent Spike changes to some known wastewater sequences.
For example: https://github.com/cov-lineages/pango-designation/issues/451
corneliusroemer
commented
2 years ago @cvejris I have explained above that the placement by Usher is not reliable in the case of convergent evolution and that S:460K is more likely to be homoplasic than E:S55F. It's similar to BA.2.3.20 but in the same way that bats and birds are similar despite having evolved flight independently. Of course here this is hard to prove but it's most likely the case.
cvejris
commented
2 years ago @corneliusroemer I know, but what about the combination of 460K with reversion of R493? These are both rare in BA.2 in general, so Usher lumping them together without phylogenetic relatedness seems to me less likely than that they represent genuine, monophyletic branch in BA.2.3 diversity
c19850727
commented
2 years ago An important information from NIID's latest variant report:
 https://www.niid.go.jp/niid/ja/2019-ncov/2551-cepr/11469-sars-cov-2-20.html
https://www.niid.go.jp/niid/ja/2019-ncov/2551-cepr/11469-sars-cov-2-20.html
"These 3 cases were on different flights arrived on different dates, and no epidemiological link has been established among them."
 ItokawaK
commented
2 years ago
ItokawaK
commented
2 years ago @corneliusroemer @alurqu @TakaKen6
I should note that threre is a genome labeled with BA.2.3.2 which share some S profile Y144-, L452R and the R493Q reversion.
| Virus name: | hCoV-19/Vietnam/NHTD-OUCRU3027/2022 |
| Accession ID: | EPI_ISL_13358783 |
| Collection date: | 2022-05-13 |
| Location: | Asia / Vietnam / Hanoi |
| Submitter: | Nguyen, Trang |
| Submission Date: | 2022-06-20 |
A sub-tree like below was only obitained when the 7039th site was masked, though.
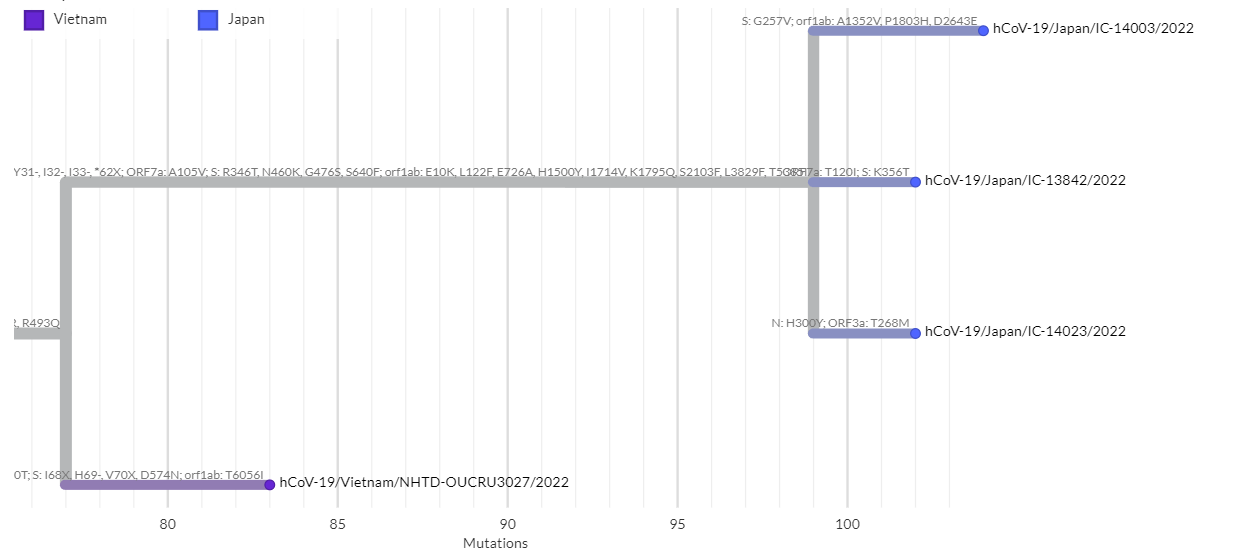
If hCoV-19/Vietnam/NHTD-OUCRU3027/2022 and the above three genomes really share a close common ancestor, S: N460K occured after S: L452R and the S: R493Q reversion.
 TakaKen6
commented
2 years ago
TakaKen6
commented
2 years ago We have found BA.2.3.2 with 7 extra S1 mutations again in one entrant from Vietnam/South Korea. Virus name: hCoV-19/Japan/IC-14136/2022 Accession ID: EPI_ISL_14906835
FedeGueli
commented
2 years ago One more sequence from Singapore, local case, i suggest to fast designate this one: EPI_ISL_14917719
corneliusroemer
commented
2 years ago Thanks @TakaKen6 and @FedeGueli - I've designated this lineage as BS.1 (BA.2.3.2.1)
c19850727
commented
1 year ago EPI_ISL_14975762, local case from Korea, collected on Aug 31st.
corneliusroemer
commented
1 year ago And intermediate from Indonesia was just uploaded

hCoV-19/Indonesia/JK-CW62-2369/2022|EPI_ISL_15005560|2022-08-20


Description
Sub-lineage of:BA.2.3.2 Earliest sequence: 2022-08-20 Most recent sequence: 2022-08-23 Countries circulating: Vietnam
Genomes hCoV-19/Japan/IC-13842/2022, EPI_ISL_14853710 hCoV-19/Japan/IC-14003/2022, EPI_ISL_14853827 hCoV-19/Japan/IC-14023/2022, EPI_ISL_14853841
Evidence
Despite the BA.5 Omicron variant recently becoming prevalent globally, we identified the other variant, BA.2.3.2-positive three entrants from Vietnam (Pango lineage3, version: 4.1.2), although the genome sequences exhibited additional mutation profiles: IC-13842 (collection date: 8/20/2022; GISAID ID: EPI_ISL_14853710), IC-14023 (collection date: 8/22/2022; GISAID ID: EPI_ISL_14853841), and IC-14003 (collection date: 8/23/2022; GISAID ID: EPI_ISL_14853827). The figure shows the number of BA.2.3.2 sequences deposited in GISAID from each country.
BA.2.3.2 in all subjects carried more than 90 nucleotide mutations that have not previously been detected in this variant (typical mutation frequency: 60–70) (Table), and shared many mutations, especially 1) mutations leading to common amino acid changes in the Spike protein, namely, Y144del, a three-amino-acid insertion at position 212 (ins_S:212:NGE), R346T, L452R, N460K, G476S, R493Q (reversion), and S640F; and 2) a frame shift by a deletion in open reading frame 6 (ORF6) (27266_27300del).
Phylogenic placement by UShER (tree version: 2022-09-05) supported monophyly of those genomes and a long branch from the other BA.2.3.2 genomes known to date (A and B).