 piechottam
commented
7 years ago
piechottam
commented
7 years ago Hi,
I am doing RDDs with JACUSA (working great !)
Okay, I assume your JACUSA call is something like this:
java jacusa.jar call-2 -a H:1 DNA.bam RNA.bam
My test statistics scores range form 0.001 - 300. Is this score meaningful when working without replicates?
What would be a descent/acceptable minimum (10, 100, 200)?
We tested JACUSA in the RDD scenario with replicates and WITHOUT replicates.
In your case you can add -T 1.15 to your jacusa call. This is an empirically derived threshold for the RDD scenario without any replicates.
Check the supplement (3.5 Derived threshold in [...]) https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1432-8 ,
if you like.
In the manual you mention 'base IJ columns indicate inverted base count if on >negative >strand’. In this case, is the vector (A,C,G,T) inverted for RNA sample on minus strand as >(T,G,C,A)? Is the following example correctly interpreted for the minus strand ('115' corresponds to >C or G)?
This depends on your library and on the employed JACUSA version:
Check 3.1.1 Strand information in the updated manual https://github.com/dieterich-lab/JACUSA/blob/master/manual/manual.pdf
Prior JACUSA 1.2:
Your JACUSA call would have an option such as: -P U,S => unstraded DNA and Stranded RNA.
If you have a lib where the first read is sequenced -> you have to invert the base counts. (you can use JacusaHelper for that.)
If you have a lib where the second read is sequenced -> you don't have to do anything.
As of version 1.2, JACUSA supports stranded paired end reads.
The format of -P has changed. The options are borrowed from tophat: FR-FIRSTSTRAND, FR-SECONDSTRAND and UNSTRANDED.
Your option could be -P UNSTRANDED,FR-FIRSTSTRAND if that corresponds to your libs. The base counts will be correctly inverted corresponding to library and strand.
Hope, that helps,
Best,
Michael
 u9090
u9090 bostanict
bostanict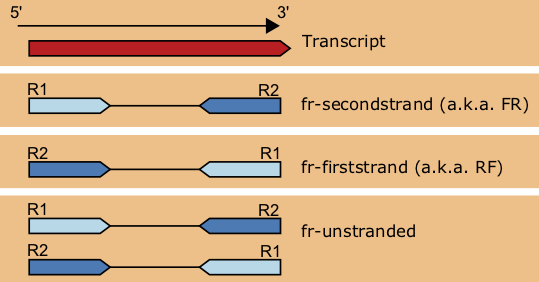{kind=link}
Hi,
I am doing RDDs with JACUSA (working great !)
My test statistics scores range form 0.001 - 300. Is this score meaningful when working without replicates? What would be a descent/acceptable minimum (10, 100, 200)?
In the manual you mention 'base IJ columns indicate inverted base count if on negative strand’. In this case, is the vector (A,C,G,T) inverted for RNA sample (FR-FIRSTSTRAND) on minus strand as (T,G,C,A)? Is the following example correctly interpreted for the minus strand ('115' corresponds to C or G)?
About the vcf output, does the ALT base reported is the one with the highest number of reads in samples 2 (after the REF ones)?
Thanks !