 jiajic
commented
1 year ago
jiajic
commented
1 year ago Hi @ayumatsubo, sorry for the delay in our reply.
How to best normalize the data from CosMx with their provided negative probe information is definitely an interesting question, and something that we are trying to work out as well.
It might not be expected that there would be a major change in the overall expression matrix when subtracting the mean negative probe detections/cell and then using ceiling(). There are a total of 20 negative probes, and the distribution of negative probe detections across all cells in Lung 12 FOVs 2-4 (8066 cells) reveals that very few contain at least the 20 detections that would be necessary to decrease raw expression matrix value by 1.
When looking at the data, the number of cells where length(negmean[negmean >= 1]) was only 3.
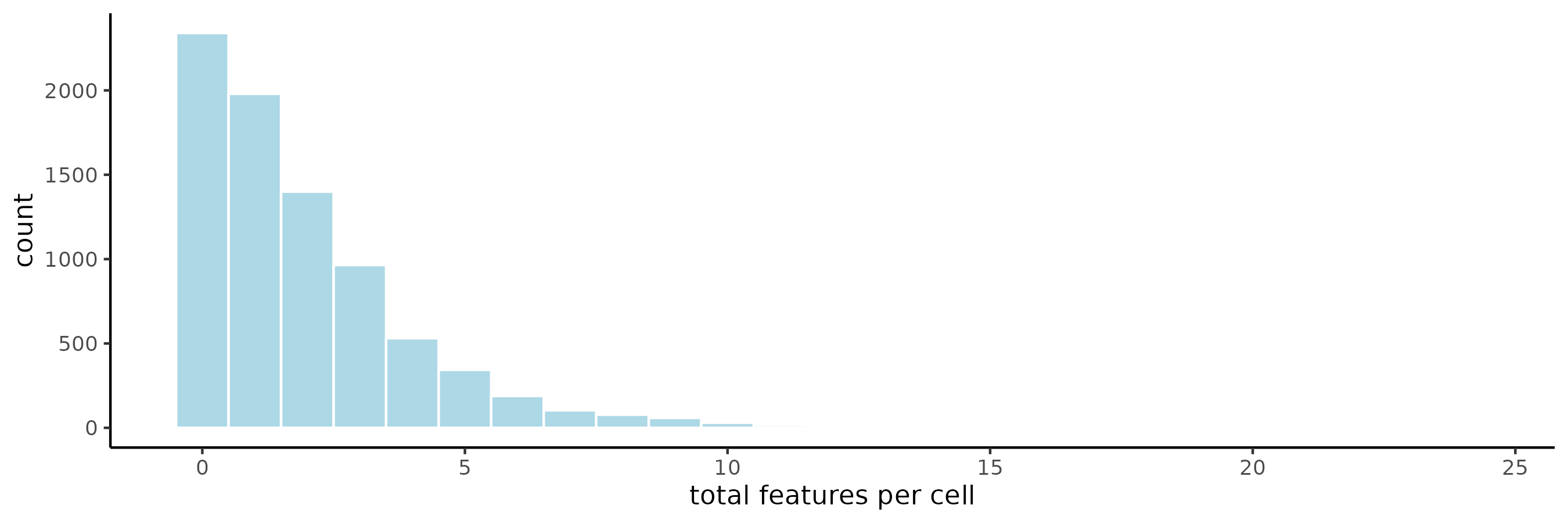
The following is a script I used to look into this:
In Giotto Suite 3.1, createGiottoCosMxObject() will generate a gobject with the rna expression and negative probe detections separated into independent feature types ('rna' and 'neg_probe' respectively), meaning that after running overlap, they will generate separate aggregate cell by count matrices.
The following code should work for gobject creation, extracting the matrix information, and setting it back after your modifications.
# obj creation
fov_join = createGiottoCosMxObject(cosmx_dir = data_path,
data_to_use = 'subcellular',
FOVs = c(2,3,4))
# Generate overlaps and expr mats
fov_join = calculateOverlapRaster(fov_join, feat_info = 'rna')
fov_join = calculateOverlapRaster(fov_join, feat_info = 'neg_probe')
fov_join = overlapToMatrix(fov_join, feat_info = 'rna')
fov_join = overlapToMatrix(fov_join, feat_info = 'neg_probe')
# ** Note: `counts_all[]` is shorthand for `counts_all@exprMat` **
# extract the raw matrix
counts_feat <- Giotto:::get_expression_values(fov_join, feat_type = 'rna')
counts_neg <- Giotto:::get_expression_values(fov_join, feat_type = 'neg_probe')
# calculate the mean of negative probe per cell
negmean <- Matrix::colMeans(counts_neg[])
# subtract negmean
count_replace <- t(apply(counts_feat[],1,function(row) row - negmean))
count_replace[count_replace < 0 ] <- 0
count_replace <- ceiling(count_replace)
# replace the matrix
counts_feat[] <- as(count_replace, 'dgCMatrix')
fov_join <- Giotto:::set_expression_values(fov_join, values = counts_feat) ayumatsubo
ayumatsubo
I'm trying to correct the negative probe value to suppress the background noise in CosMx dataset.
I calculated the mean count value of negative probe per cell and subtract the mean value from raw count matrix, and then if raw count data < 0, I replace it to 0 and build new corrected count matrix.
My question is how can I replace the original raw count matrix to count matrix.
I tried to use
set_expression_valuefunction, but the result seems to be the same.My code is following from correcting raw count matrix to replacing the raw count data.
#extract the raw matrixcounts_all <- fov_join@expression$cell$rna$raw#calculate the mean of negative probe per cellnegmean <- Matrix::colMeans(counts_all[rownames(counts_all)[grepl('^Neg',rownames(counts_all))],])#subtract negmeancount_replace <- t(apply(counts_all,1,function(row) row - negmean))count_replace[count_replace < 0 ] <- 0count_replace <- ceiling(count_replace)# replace the matrixfov_join <- set_expression_values(fov_join, name = 'raw', values = as(count_replace, 'dgCMatrix')) # replace the raw matrixThank you for advance.