 mourisl
commented
2 years ago
mourisl
commented
2 years ago Thanks for sharing your test results with us. Indeed, the length distribution change is too sharp, and the fragment length 0 should not happen either. Is the data publicly available? If it is not, could you please share a few hundreds of the paired-end sequence data with us (no need for the barcode file)? We will look into this issue. Thank you.
 loraince
loraince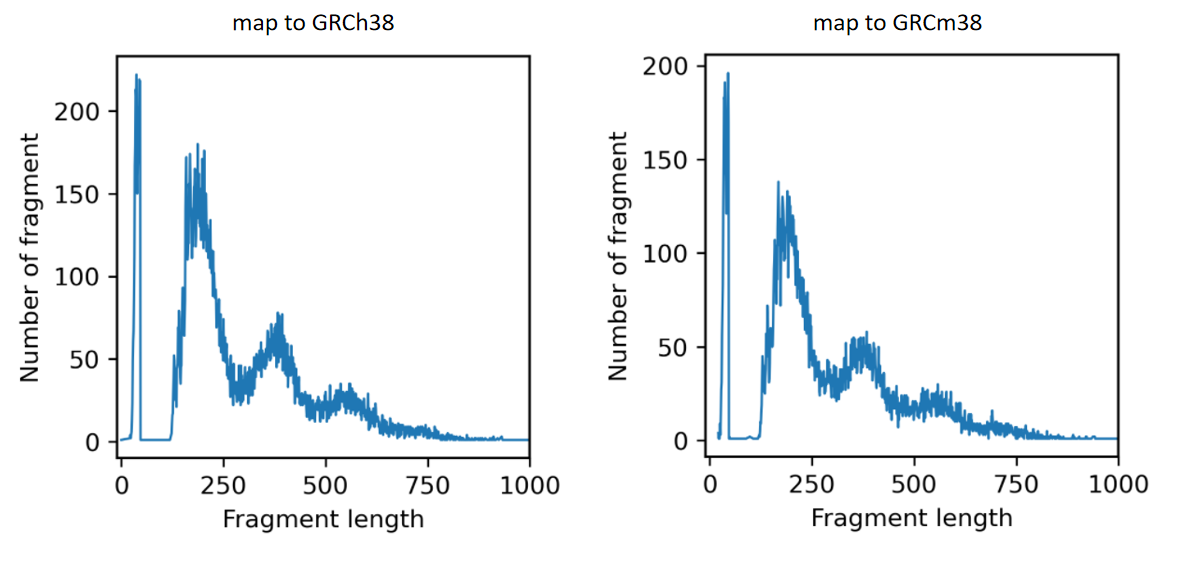
 haowenz
haowenz
Chromap is indeed an ultrafast tool. Thanks for developing and this method. However, I have encountered two problems when I using chromap with scATAC-seq data.
The first one is that when I analyzed the distribution of fragment length, I found a dramatic loss of fragments between 47bp-120bp The results are shown in these figures.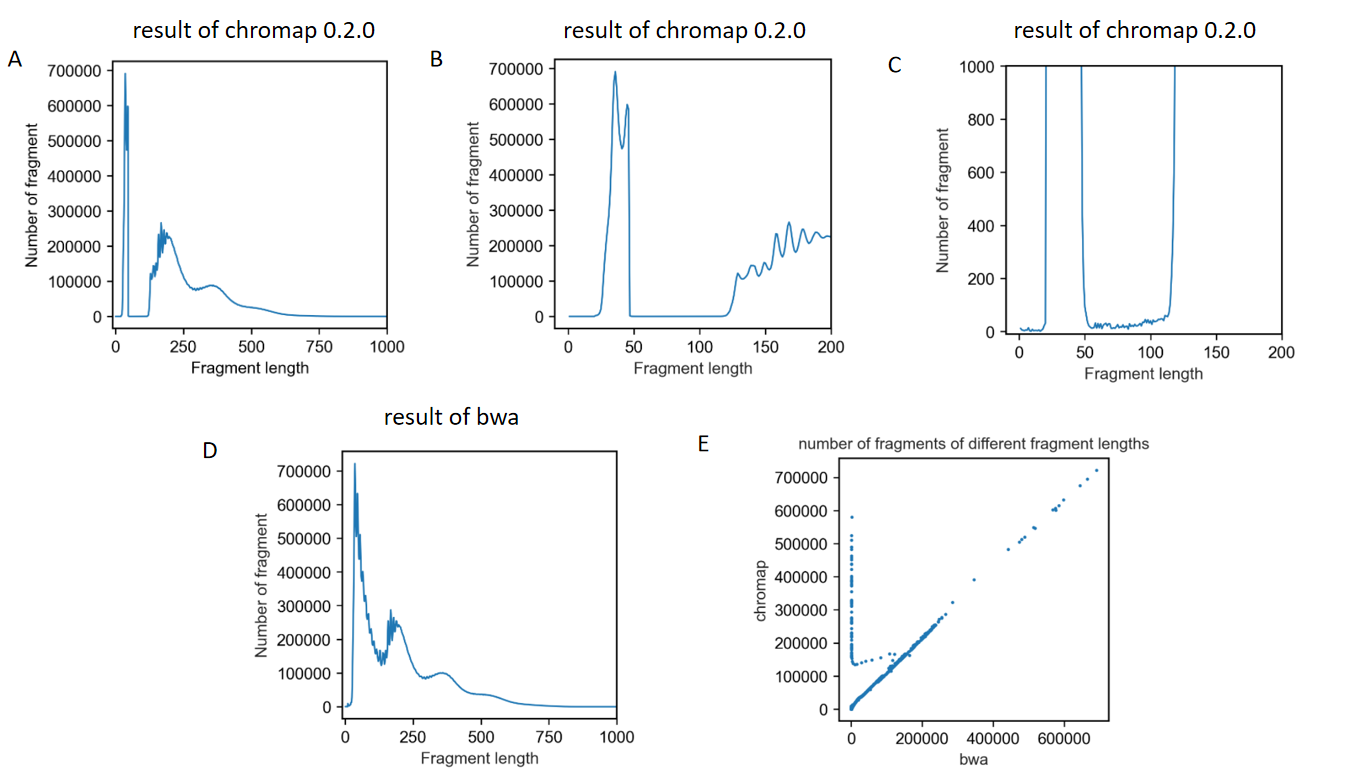
Another problem is that fragments of 0bp existed in the result bed file.
These two problems existed for all data of my different experiments. All these data have 10x-like scATAC-seq structures, using 150bp paired end sequencing.
The command was like this: chromap --preset atac -x ${index} -r ${fasta} -1 ${inputR1} -2 ${inputR3} -o ${output} -b ${inputR2} --barcode-whitelist ${whitelist}
I also tried without the barcode fastq, running like this: chromap --preset atac -x ${index} -r ${fasta} -1 ${inputR1} -2 ${inputR3} -o ${output}
However, the two problems still existed for my data.
When I tried another scATAC-seq dataset with 50bp paired end sequencing, there two problems did not appear.