 mgavery
commented
4 years ago
mgavery
commented
4 years ago 
Closed mgavery closed 3 years ago
mgavery
commented
4 years ago
mgavery
commented
4 years ago 
mgavery
commented
4 years ago This is the file with data in screenshots above: MethylKit_various_coverage.xlsx
 shellywanamaker
commented
4 years ago
shellywanamaker
commented
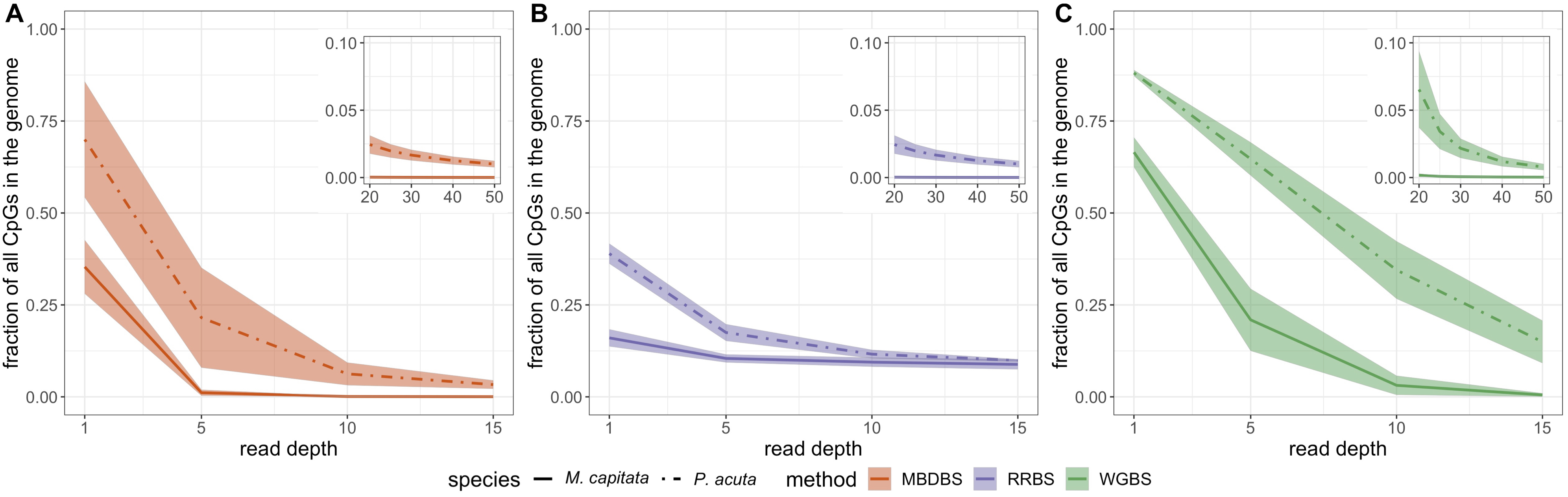
4 years ago Interesting, so it seems more coverage leads to higher correlations of biological replicates but lower correlations across methods?
mgavery
commented
4 years ago Yes, I saw that pattern too. Differences are slight though, I would say.
The PCAs are interesting too. Method differences become more pronounced at higher coverage
mgavery
commented
4 years ago ps. if you look at the original file I have the 5x PCAs. The default settings in MethylKit must be a little different from what you used @shellytrigg , because the separation is a little different, but generally speaking the trends are the same.
shellywanamaker
commented
4 years ago the separation does look different. @mgavery in your original file are the 5x PCAs generated with the '5x mincov' or '10x mincov' (https://github.com/hputnam/Meth_Compare/issues/87)? Discrepancies could also be from differences in scaling and centering so I can play around with that
mgavery
commented
4 years ago @shellytrigg, all PCAs were generated by modifying the "mincov" parameter in the "methRead" fxn. I confirmed the # of 5x loci for these PCAs and they matched. I bet it's scaling diffs, etc.
So the PCA on the far right in the file for 5x was generated using this code (Mcap example): myobj_Mcap_all_TG=methRead(Mcap_all_TG.list, sample.id = list("WGBS 1","WGBS 2","WGBS 3","MBDBS 1","MBDBS 2","MBDBS 3","RRBS 1","RRBS 2","RRBS 3"), assembly = "Mcap_genome", treatment = c(0,0,0,0,0,0,1,1,1), context = "CpG", pipeline = "bismarkCoverage", mincov = 5)
 DrK-Lo
commented
4 years ago
DrK-Lo
commented
4 years ago Thank you for doing this cool analysis! Hmm, this didn't match my intuition at all. I would have thought that with higher coverage, the individuals would have started to cluster together more and the methods would have been less clustered. Can you describe for me the dataframe that goes into the correlations and PC plots? Is it CpGs in rows, samples in columns, with the estimated methylation level in the cells?
mgavery
commented
4 years ago The PCAs and correlations were done in methylKit, which is a little 'behind the scenes', but, yes, the main dataframe frame that methylKit works with is CpGs in rows, samples in columns and %methylation (coverage C/total coverage) in the cells
DrK-Lo
commented
4 years ago Thanks that make sense. This pattern must be driven by the higher number of DML among methods at higher coverage. For the loci that are >50% different between the library prep methods, is there any trend for one library prep method to have higher or lower methylation levels than another?
mgavery
commented
4 years ago Particularly for P.act, MBD is giving consistently higher calls than WGBS or RRBS. You can see this in the correlation plots in Figure 6. Less obvious trends for Mcap and for the other comparisons
mgavery
commented
4 years ago @shellytrigg , here are the %meth files from methylKit at various coverage cutoffs. Number of loci changes quite dramatically as you can imagine :)
number of lines: 73 730 11807 Mcap_percmeth_100x.txt 1862 18620 253024 Mcap_percmeth_10x.txt 332 3320 52499 Mcap_percmeth_50x.txt 4667 46670 537721 Mcap_percmeth_5x.txt 2007 20069 357034 Pact_percmeth_100x.txt 33592 335919 4850220 Pact_percmeth_10x.txt 5160 51599 888539 Pact_percmeth_50x.txt 93715 937149 11027854 Pact_percmeth_5x.txt Mcap_percmeth_100x.txt Mcap_percmeth_50x.txt Mcap_percmeth_10x.txt Mcap_percmeth_5x.txt Pact_percmeth_100x.txt Pact_percmeth_50x.txt Pact_percmeth_10x.txt Pact_percmeth_5x.txt
shellywanamaker
commented
4 years ago awesome, thanks @mgavery! will post the PCAs as soon as I make them
mgavery
commented
3 years ago PCAs performed using same method as supp material. Same x and y scales for all


shellywanamaker
commented
3 years ago Nice @mgavery ! So the individuals do get closer and closer with higher coverage when all PCAs are on the same scale. And MBD methylomes are the least similar, would you say?
shellywanamaker
commented
3 years ago just wanted to note that methylkit does centering and scaling (lines 150-214, source code: https://github.com/al2na/methylKit/blob/master/R/clusterSamples.R)
shellywanamaker
commented
3 years ago I've plotted the PCAs as 2 rows and 4 columns and added it to the manuscript as the new main text figure 4.
Here's where this new figure lives: https://github.com/hputnam/Meth_Compare/blob/master/output/figures/Fig_4.jpg
Here's the code: https://github.com/hputnam/Meth_Compare/blob/master/code/02.01-5xUnionBedCpGs_PCA.Rmd
Now just need to update the main text to match the new figure so I'll make a new issue for that.
{kind=link}
Katie's comment: think this should be a main figure in the manuscript, at least I think it is more important than Fig 1 and Fig 3. I also disagree with this sentence. Since you filtered to loci with 5x in common across all samples, what this tells you is that when methods do not cluster together in PC space, there are fundamental differences in methylation calls across methods (at least when methylation level is based on a minimum of 5x - would be good to increase this to 50x and see if differences are minimized). The way it is written implies that it has something to do with sequencing to saturation, but since the data was filtered to the same set of CpGs across methods, this cannot explain the observed pattern (unless I am misunderstanding what is meant by sequencing to saturation)
@mgavery:make PCAs and corr plots in MethylKit with 5x, 10x, 50x and 100x coverage and put them here for us all to look at.