 fpusan
commented
7 months ago
fpusan
commented
7 months ago Try adding --assembler_options "useGrid=false" when running SqueezeMeta.pl, or changing the assembler from canu to flye.
Let me know if it works.
Closed vmevada102 closed 6 months ago
fpusan
commented
7 months ago Try adding --assembler_options "useGrid=false" when running SqueezeMeta.pl, or changing the assembler from canu to flye.
Let me know if it works.
 vmevada102
commented
7 months ago
vmevada102
commented
7 months ago Adding the -a canu not helped me to process.
The following command processed., SqueezeMeta.pl -m sequential -s '/home/vishal/Desktop/squeezemetarun/run_ion3.samples' -f '/home/vishal/Desktop/squeezemetarun/raw' -t 24 -miniden 20 --minion --force_overwrite -a canu
fpusan
commented
7 months ago I didn't tell you to add -a canu. Try removing --minion and adding -a flye.
vmevada102
commented
7 months ago Command
SqueezeMeta.pl -m sequential -s '/home/vishal/Desktop/squeezemetarun/run_ion3.samples' -f '/home/vishal/Desktop/squeezemetarun/raw' -t 24 -miniden 20 -a flye
output
mkdir: cannot create directory ‘/home/vishal/Desktop/squeezemetarun’: File exists
SqueezeMeta v1.6.0, September 2022 - (c) J. Tamames, F. Puente-Sánchez CNB-CSIC, Madrid, SPAIN
Please cite: Tamames & Puente-Sanchez, Frontiers in Microbiology 9, 3349 (2019). doi: https://doi.org/10.3389/fmicb.2018.03349
Run started Thu Nov 16 15:10:20 2023 in sequential mode 1 metagenomes found: P091
--- SAMPLE P091 -------- Now creating directories Reading configuration from /home/vishal/Desktop/squeezemetarun/P091/SqueezeMeta_conf.pl [0 seconds]: STEP1 -> RUNNING ASSEMBLY: 01.run_all_assemblies.pl (flye) Working for sample P091 Preparing files for pair1: cat /home/vishal/Desktop/squeezemetarun/raw/SRR12481289.fastq.gz > /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/par1.fastq.gz Running assembly with flye: perl /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/lib/SqueezeMeta/assembly_flye.pl /home/vishal/Desktop/squeezemetarun/P091 P091 /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/par1.fastq.gz /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/ mv: cannot stat ‘/home/vishal/Desktop/squeezemetarun/P091/data/flye/assembly.fasta’: No such file or directory Error running command: /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/bin/Flye-2.9/bin/flye -o /home/vishal/Desktop/squeezemetarun/P091/data/flye --plasmids --meta --genome-size 2.6g --threads 24 --nano-raw /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/par1.fastq.gz > /home/vishal/Desktop/squeezemetarun/P091/syslog 2>&1; mv /home/vishal/Desktop/squeezemetarun/P091/data/flye/assembly.fasta /home/vishal/Desktop/squeezemetarun/P091/data/flye/contigs.fasta at /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/lib/SqueezeMeta/assembly_flye.pl line 36. Running prinseq (Schmieder et al 2011, Bioinformatics 27(6):863-4) for selecting contigs longer than 200
ERROR: could not find input file "/home/vishal/Desktop/squeezemetarun/P091/results/01.P091.fasta".
Try 'perl prinseq-lite.pl -h' for more information. Exit program. Error running command: /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/bin/prinseq-lite.pl -fasta /home/vishal/Desktop/squeezemetarun/P091/results/01.P091.fasta -min_len 200 -out_good /home/vishal/Desktop/squeezemetarun/P091/results/prinseq; mv /home/vishal/Desktop/squeezemetarun/P091/results/prinseq.fasta /home/vishal/Desktop/squeezemetarun/P091/results/01.P091.fasta > /dev/null 2>&1 at /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/scripts/01.run_all_assemblies.pl line 242. Stopping in STEP1 -> 01.run_all_assemblies.pl. Program finished abnormally
If you don't know what went wrong or want further advice, please look for similar issues in https://github.com/jtamames/SqueezeMeta/issues Feel free to open a new issue if you don't find the answer there. Please add a brief description of the problem and upload the /home/vishal/Desktop/squeezemetarun/P091/syslog file (zip it first) Died at /home/vishal/miniconda3/envs/squeezemeta/bin/SqueezeMeta.pl line 921.
fpusan
commented
7 months ago Maybe your input sequences are too short for flye. You can also try with the default assembler (megahit) or with spades. Also remove the output directories from the previous run before launching again. Let me know
vmevada102
commented
7 months ago No, length of sequences are around morethan 300-400 bps.
Spades as well megahit no working for these samples.
Tool is perfectly working for another samples from Illumina.
Command
SqueezeMeta.pl -m sequential -s '/home/vishal/Desktop/squeezemetarun/run_ion3.samples' -f '/home/vishal/Desktop/squeezemetarun/raw' -t 24 -miniden 20 -a spades
Output (squeezemeta) [vishal@vnsgu squeezemetarun]$ SqueezeMeta.pl -m sequential -s '/home/vishal/Desktop/squeezemetarun/run_ion3.samples' -f '/home/vishal/Desktop/squeezemetarun/raw' -t 24 -miniden 20 -a spades mkdir: cannot create directory ‘/home/vishal/Desktop/squeezemetarun’: File exists
SqueezeMeta v1.6.0, September 2022 - (c) J. Tamames, F. Puente-Sánchez CNB-CSIC, Madrid, SPAIN
Please cite: Tamames & Puente-Sanchez, Frontiers in Microbiology 9, 3349 (2019). doi: https://doi.org/10.3389/fmicb.2018.03349
Run started Thu Nov 16 15:28:59 2023 in sequential mode 1 metagenomes found: P091
--- SAMPLE P091 -------- Now creating directories Reading configuration from /home/vishal/Desktop/squeezemetarun/P091/SqueezeMeta_conf.pl [0 seconds]: STEP1 -> RUNNING ASSEMBLY: 01.run_all_assemblies.pl (spades) Working for sample P091 Preparing files for pair1: cat /home/vishal/Desktop/squeezemetarun/raw/SRR12481289.fastq.gz > /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/par1.fastq.gz Running assembly with spades: perl /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/lib/SqueezeMeta/assembly_spades.pl /home/vishal/Desktop/squeezemetarun/P091 P091 /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/par1.fastq.gz /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/ Error running command: /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/bin/SPAdes/spades.py --meta --pe1-1 /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/par1.fastq.gz --pe1-2 /home/vishal/Desktop/squeezemetarun/P091/data/raw_fastq/ -m 400 -k 21,33,55,77,99,127 -t 24 -o /home/vishal/Desktop/squeezemetarun/P091/data/spades >> /home/vishal/Desktop/squeezemetarun/P091/syslog 2>&1 at /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/lib/SqueezeMeta/assembly_spades.pl line 39. Running prinseq (Schmieder et al 2011, Bioinformatics 27(6):863-4) for selecting contigs longer than 200
ERROR: could not find input file "/home/vishal/Desktop/squeezemetarun/P091/results/01.P091.fasta".
Try 'perl prinseq-lite.pl -h' for more information. Exit program. Error running command: /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/bin/prinseq-lite.pl -fasta /home/vishal/Desktop/squeezemetarun/P091/results/01.P091.fasta -min_len 200 -out_good /home/vishal/Desktop/squeezemetarun/P091/results/prinseq; mv /home/vishal/Desktop/squeezemetarun/P091/results/prinseq.fasta /home/vishal/Desktop/squeezemetarun/P091/results/01.P091.fasta > /dev/null 2>&1 at /home/vishal/miniconda3/envs/squeezemeta/SqueezeMeta/scripts/01.run_all_assemblies.pl line 242. Stopping in STEP1 -> 01.run_all_assemblies.pl. Program finished abnormally
If you don't know what went wrong or want further advice, please look for similar issues in https://github.com/jtamames/SqueezeMeta/issues Feel free to open a new issue if you don't find the answer there. Please add a brief description of the problem and upload the /home/vishal/Desktop/squeezemetarun/P091/syslog file (zip it first) Died at /home/vishal/miniconda3/envs/squeezemeta/bin/SqueezeMeta.pl line 921.
 jtamames
commented
7 months ago
jtamames
commented
7 months ago Hello I think the problem is that you have very few sequences, and they will not assemble no matter the assembler. You better use sqm_longreads.pl for this. Best, J
vmevada102
commented
7 months ago Thank you for your response sqm_reads.pl script working rightnow.
I am waiting for the results.
Little worried about that the result from squeezemata.pl and sql_read.pl have any resemblance or or it is different from their perspective.
can I analyse results in same way as for squeezemeta?
jtamames
commented
7 months ago You will have abundance tables as in SqueezeMeta FOr loading these into SQMtools, use loadSQMlite() instead of loadSQM() Best, J
vmevada102
commented
7 months ago BY running this command, the output folder contains following files
P09_sqm.out.allreads P09_sqm.out.allreads.funcog P09_sqm.out.allreads.funkegg P09_sqm.out.allreads.mcount P09_sqm.out.mappingstat
The results looks different. How can we get bins and other results similar to generated using squeezemeta tool.
fpusan
commented
7 months ago Hi,
Using the reads mode you can not get bins (since you are not getting an assembly, just annotating each read independently).
You can create summary tables with sqmreads2tables /path/to/project /path/to/output/tables.
At this step you can also add a query when calling the script, in order to create tables summarizing only certain taxa or functions of choice. See the documentation of sqmreads2tables.py for more details.
The summary tables can be loaded into SQMtools too, using SQM = SQMlite("/path/to/output/table. From there you can use the SQMtools functions for visualization (see the section related to SQMlite objects in this figure.
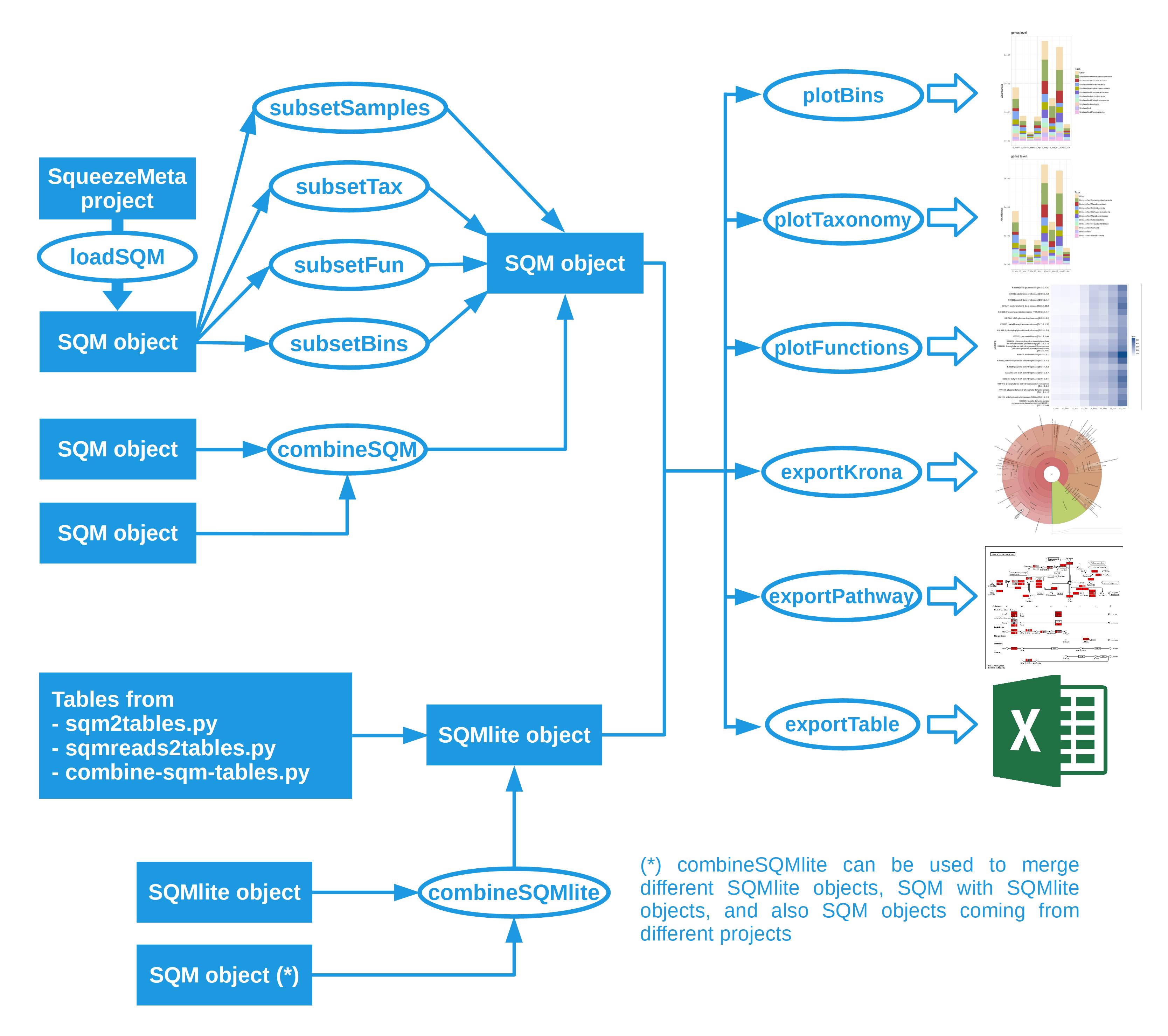{kind=link}
I am trying to run the sequences from Ion torrent S5. Unfortunately, the process terminated with the error.
Kindly guide me to resolve it.
Stopping in STEP1 -> 01.run_all_assemblies.pl. Program finished abnormally
If you don't know what went wrong or want further advice, please look for similar issues in https://github.com/jtamames/SqueezeMeta/issues Feel free to open a new issue if you don't find the answer there. Please add a brief description of the problem and upload the /home/vishal/Desktop/squeezemetarun/P09/syslog file (zip it first)
Kindly download system log from here syslog.zip