 baudrly
commented
4 years ago
baudrly
commented
4 years ago It's less dependent on the genome size per se than the fragmentation of the genome itself. If your reference genome is very fragmented (N50 < 10kb etc.) the size of the matrix will be enormous no matter the resolution and it will indeed take up a lot of memory and time. You need to preprocess your initial genome somewhat to avoid this - for instance you could run aggressive filtering so that many smaller contigs are eliminated from the scaffolding and don't clog up your RAM.
So I decreased the size of fragments contacts, which was generated from hicstuff (I used several replicates of Hi-C read files to generated contacts information at first.
What size did you use and did you try generating a matrix using restriction fragments instead?
 nadegeguiglielmoni
nadegeguiglielmoni Raina-M
Raina-M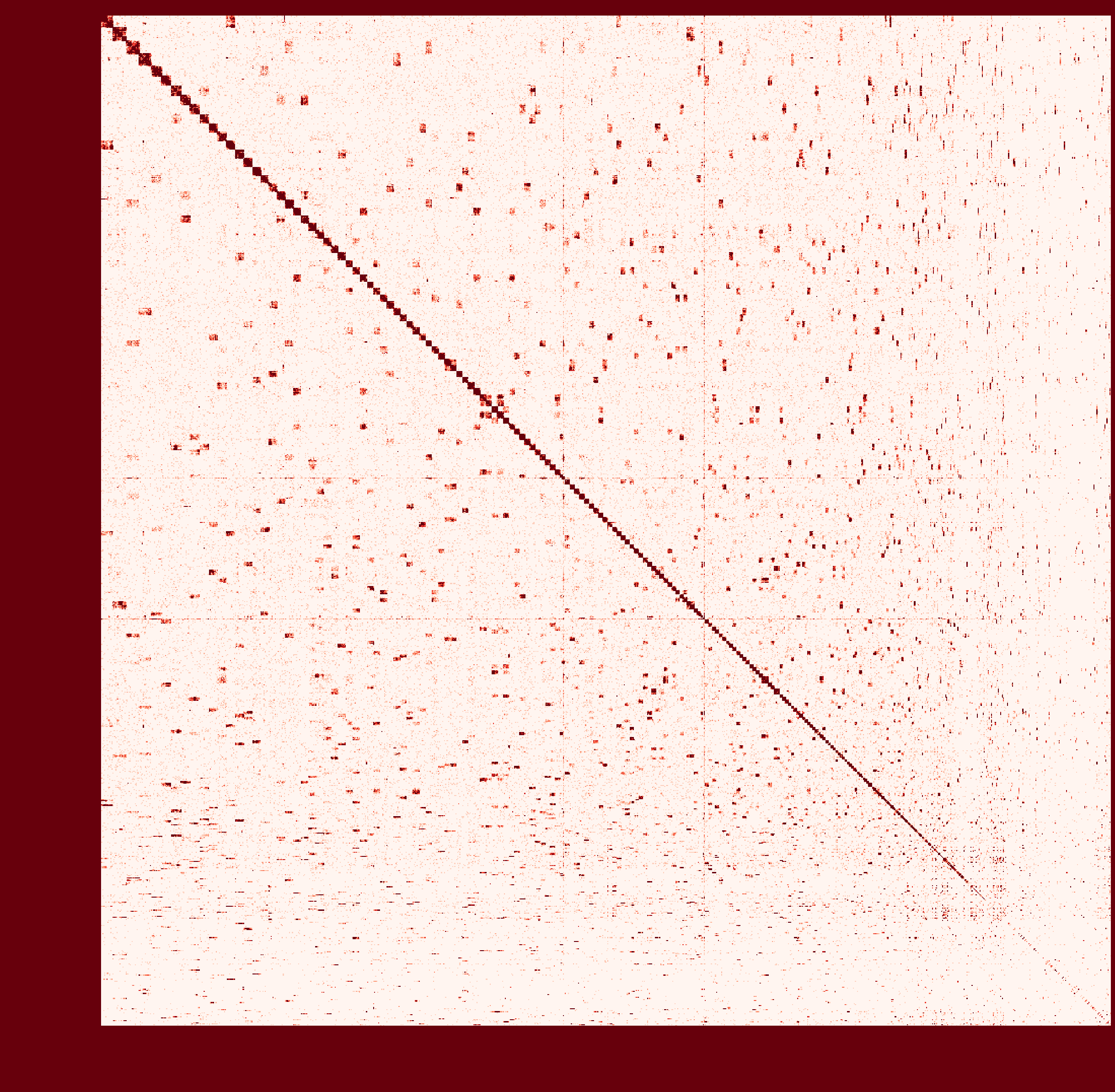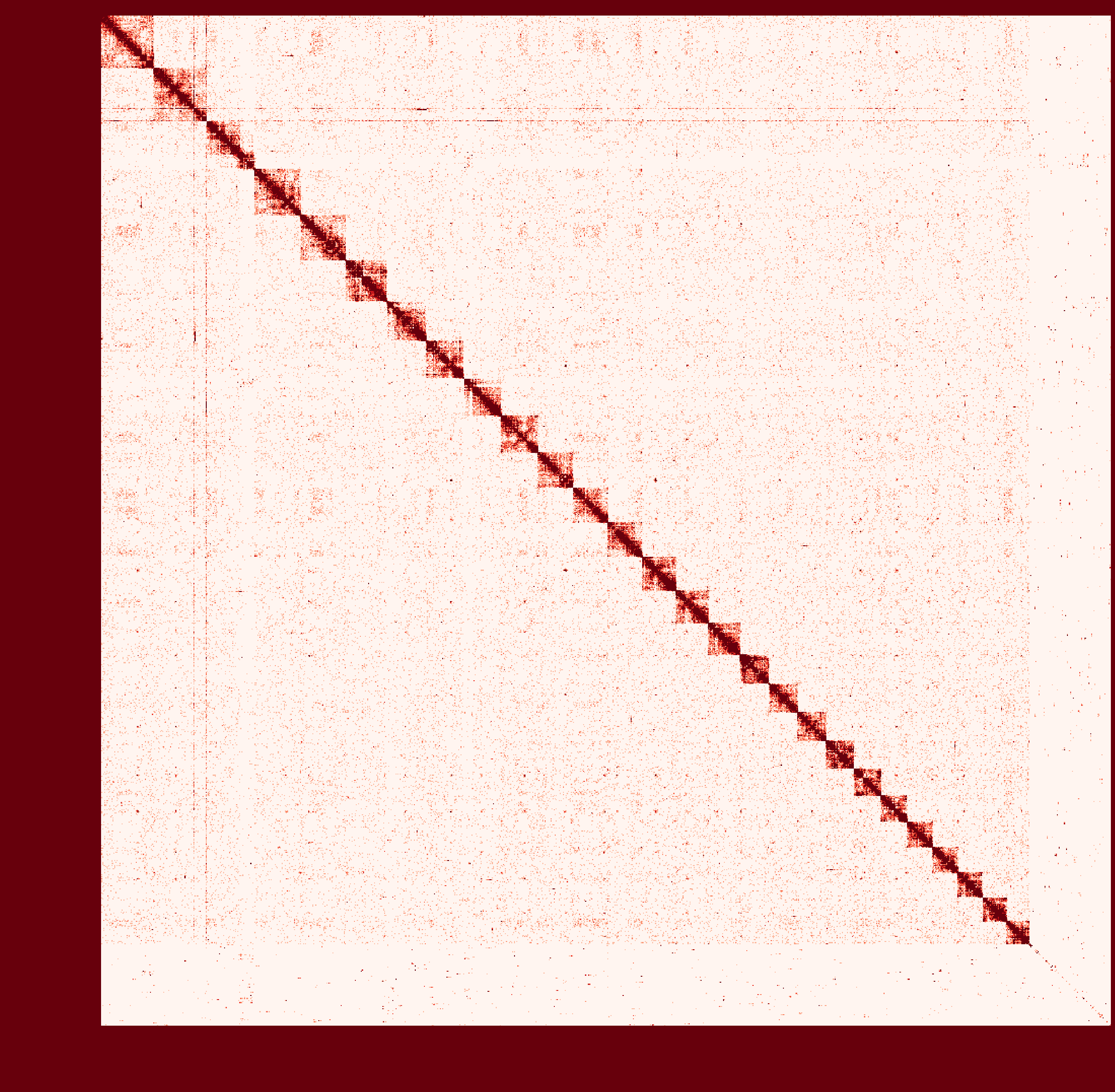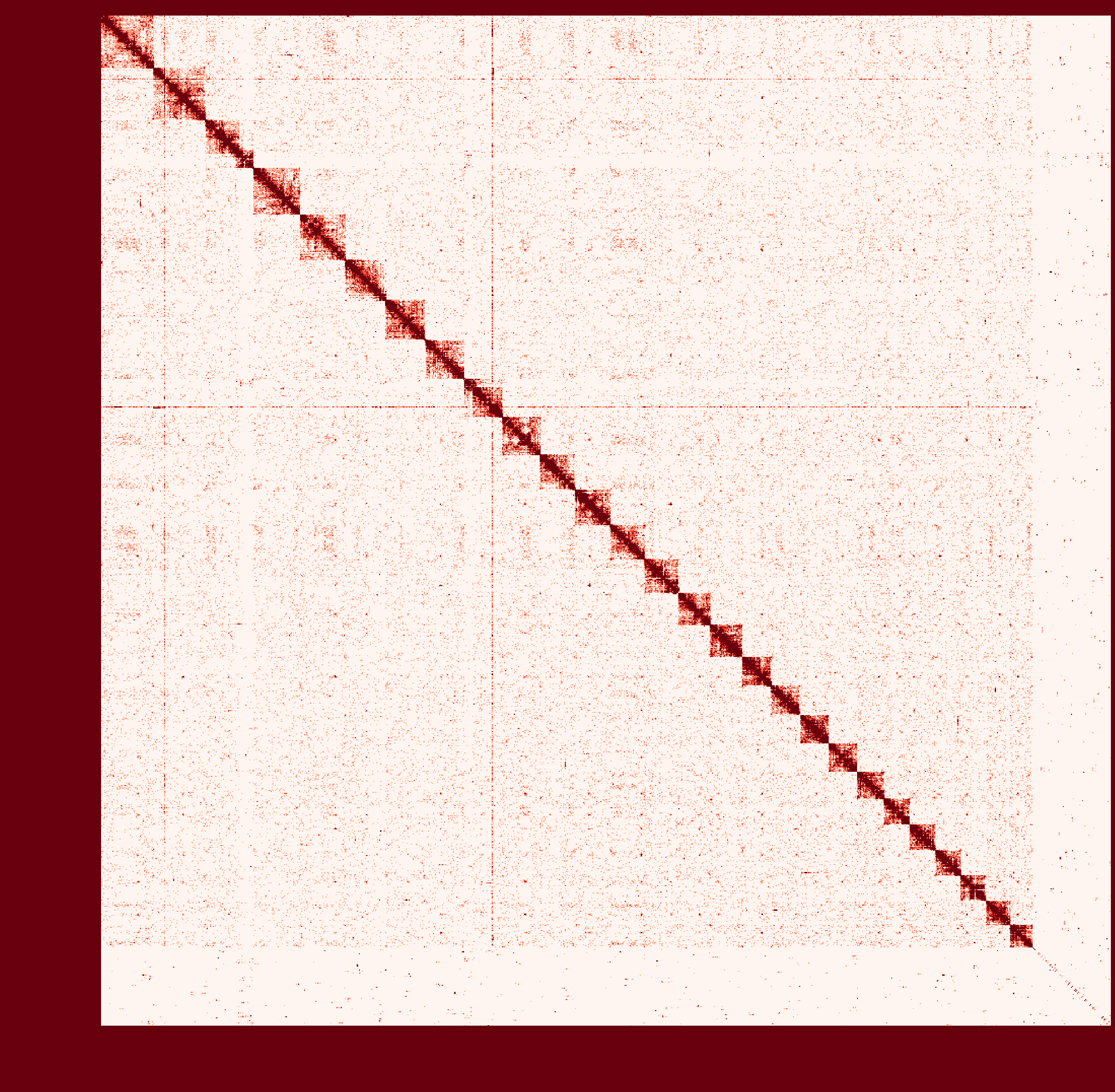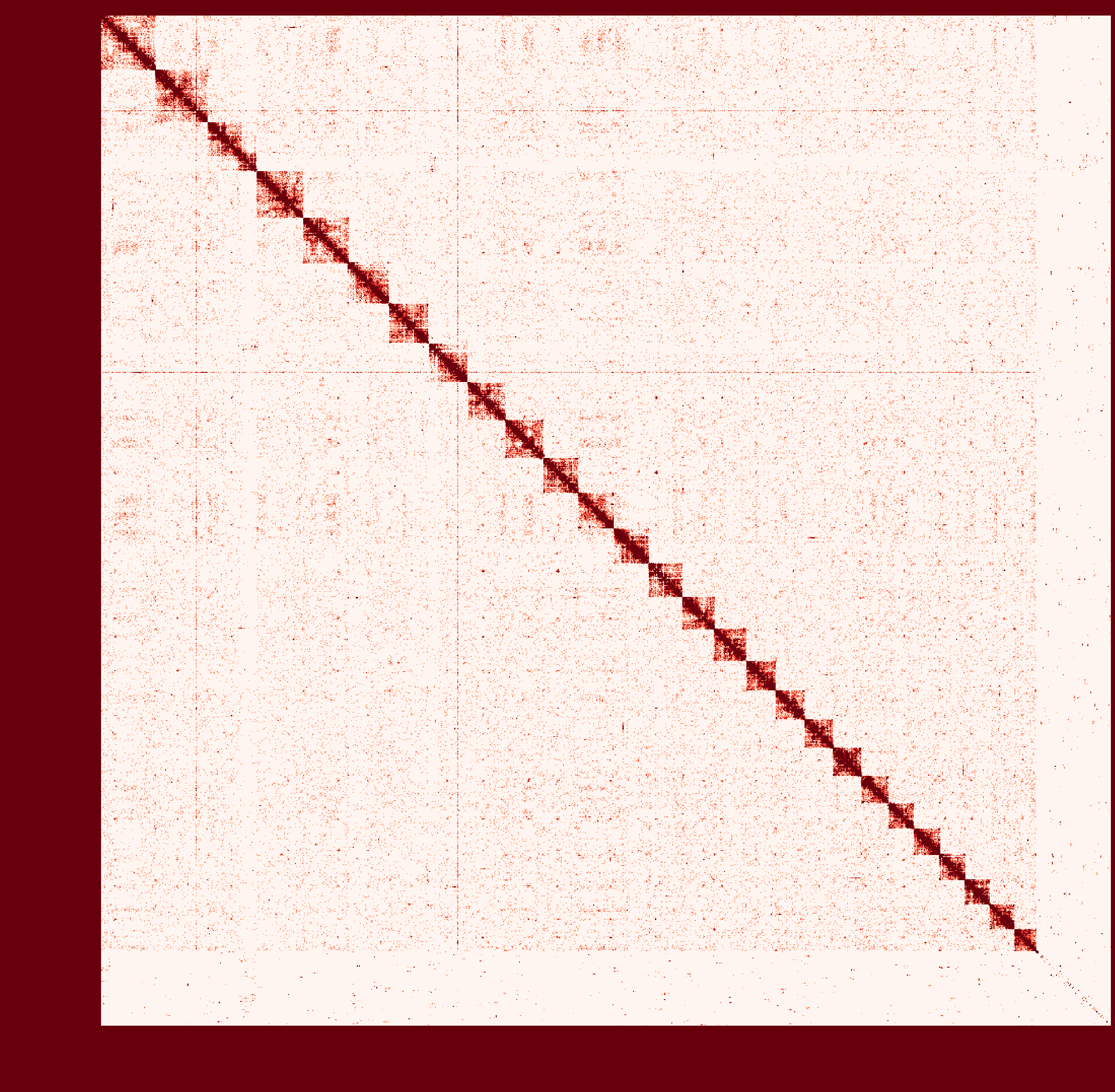
 you need to run the version with OpenGL support. You also need an X server for the movie to display, so it would probably not work on a cluster server.
you need to run the version with OpenGL support. You also need an X server for the movie to display, so it would probably not work on a cluster server.  alexcorm
alexcorm anhong11
anhong11
At first I ran a real-case sample with genome size ~2.6Gb and then
instagraalcrashed on a 400Gig machine. So I decreased the size of fragments contacts, which was generated fromhicstuff(I used several replicates of Hi-C read files to generated contacts information at first. After the machine crashed, I only used one replicate to generate smaller contact files). But still, the smaller sample needs ~30Gig Memory on the host, and 2Gig on the GPU, with 780Mabs_fragments_contacts_weighted.txt. In addition, the program ran super slowly that had ran over 1 month before killed. Before ends, it had generated new fasta file in cycles, taking approximately a day per cycle. But that's only a test sample, so I terminated it in cycle 28. Nevertheless, could you provide any information about the computational resource usage of the program. Like what I have demonstrated, the 'small' sample seems to run endless... I doubt the possibility to apply it on my real sample.