 sgibb
commented
9 years ago
sgibb
commented
9 years ago I am not quite sure that I understand your feature request completely:
- The intensity if a the matched peak is missing.
Currently calculateFragments reports just matched peaks. How could a matched peak be missing? How to get the intensity of a non-existing peak?
If 2 peaks are in the defined intervall near a theoretical peak only 1 is reported. (I guess the one with the highest intensity?) It would maybe be useful to set an option to report all matched peaks (especially informative when also the intensity is reported)
You are right. In the current implementation the highest matched peak is reported. I will think about a way to report all peaks in the user-defined tolerance range.
- Neutral losses are not considered currently (I think?).
Correct. Would it be enough to just substract the mass of NH3 respectively H2O? Or do we need a more sophisticated rule?
 adder
adder
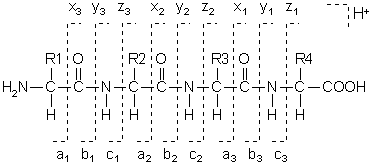
 lgatto
lgatto yafeng
yafeng smv1d14
smv1d14
Hi, I was using the calculateFragments() function in MSnbase and I think there are some small modifications that could make it more usefull.
Have a nice evening