 msbentsen
commented
3 years ago
msbentsen
commented
3 years ago Hi,
Thank you for reaching out! I will try to answer your questions in the order as listed:
- Not all TF's show nice footprints in aggregate plots - this is because not all TF's have the same ability to protect against Tn5 cleavage, not all TF's bind the DNA long enough etc. But you can still rely on the footprints.bw and .bed-files because the score is calculated as a merge of accessibility and footprint ability.
- For all three tools, you should use a merge of conditionA + conditionB narrowpeaks. You can easily make this with
$ cat condition*.narrowpeak | bedtools sort | bedtools merge > merged_peaks.bed. So even for the condition A run, the peaks should also contain the peaks from condition B to ensure that the normalization is equal. - I think the issue here is that the input
--peaksare only the unique A peaks (and it should be all peaks from both conditions, as explained in 2.). Can you try to run it with the merge of all condition peaks? This will increase the number of regions, and I also believe the bias correction will be better. You can also check the output of the bias correction in the output .pdf-file ofATACorrect. - You can benefit from smoothing the footprint if there are a lot of single-bp peaks in the footprint (e.g. few positions creating spikes in the profile). Essentially, you are taking some of the resolution away to possibly catch a more general shape. So if that works better for you, I don't see any issue in playing around with
--smooth. - The difference is that you can give several
--regions-files, which will then create subsets of the input e.g. "TFBS overlapping regions1.bed" and "TFBS overlapping regions2.bed". This is useful for checking the footprints within different sets of peaks. In your case, you could for example try--regions uniq_open_A.bed uniq_open_B.bed. The--whitelistwill simply exclude any sites not within whitelist. But you are right, that for one regions file,--regionsis technically the same as--whitelist.
I hope this helps you out!
Best, Mette
 hero-outman
hero-outman





 But I am not sure how that would influence the footprinting. I am really sorry, but I am not quite sure what to do here. The only thing I imagine is if you could share with me a subset of the reads (for example one chromosome of one condition) - then I can have a look if there might be something special not taken into account by ATACorrect. Let me know if that would be possible.
But I am not sure how that would influence the footprinting. I am really sorry, but I am not quite sure what to do here. The only thing I imagine is if you could share with me a subset of the reads (for example one chromosome of one condition) - then I can have a look if there might be something special not taken into account by ATACorrect. Let me know if that would be possible. 
Hello, msbentsen, I have some questions when using TOBIAS for my ATAC-seq data analysis, I read the docs on TOBIAS Github, but still confused. My atac-seq analysis propose is finding different TFBS motifs of A549 cell line between conditionA and conditionB; And my analysis route like:
conditionA.narrowpeakandconditionB.narrowpeak(they are whole open site of each conditon);uniq_open_in_A.bedanduniq_open_in_B.bed(they are sub-set of conditionA.narrowpeak and conditionB.narrowpeak);Next, I use TOBIAS for TF footprint analysis; after compare to your example pic, it's quite a different:
compare to your example pic, it's quite a different:
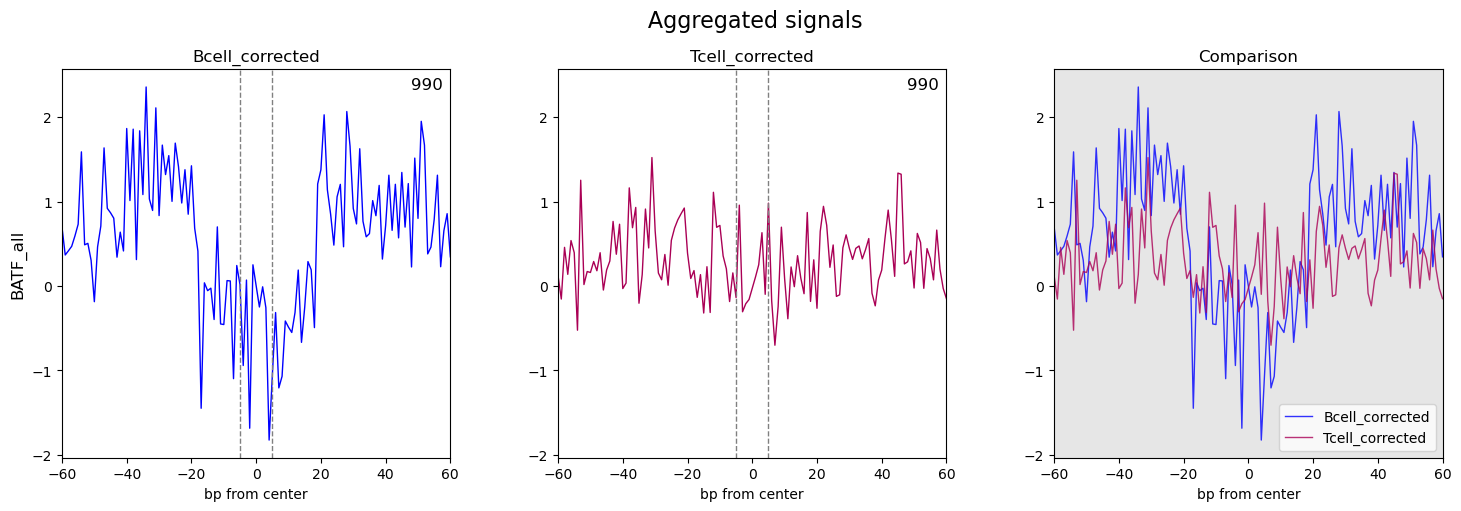
ATACorrect --> ScoreBigwig --> BINDetect --> PlotAggregate, I got my results, seems not the expected one(no obviously footprint on the aggregate plot on most enriched motif). Here are my aggregate plots:Also, I made an aggregate plot between uncorrected and corrected, still not expected(the curve shape of uncorrected and corrected are very same, just one position higher than another on the Y-axis). Here's the pic:
I tried for couple of days, still cannot get a good plot on each condition.
So I will ask some question based on the information above:
The result of scorebigwig(footprint.bw) and bindetect(TFBS.bed files) fit my expected result. But why aggregate-plot won't show a 'good-shaped' footprint plot? If aggregate-plot is not good, can I still rely on footprint.bw and TFBS.bed files?
I have conditionA and B .narrowpaek for the whole peak of two conditions, also uniq_open_A and B .bed for unique open regions(a subset of the whole peak), which kind of peak should I use in:
In aggregate plot: the plot curve between corrected.bw and uncorrected.bw, why their shape is so same, and just different position on Y-axis(not like figures in TOBIAS paper and GitHub), does this mean my correction won't work? Please see pics above; My ATACorrect code is
When plotting aggregate footprint, I try to use --smooth 3 or 5, with this param, I can get a 'better' footprint plot, does this try make sense? Here is the smoothed pic:
in
PlotAggregatemethod, What is the difference between these two arguments:--regionsand--whitelistHope you can give me some advice, thanks.