 jmichel80
commented
2 years ago
jmichel80
commented
2 years ago hi @cespos - thanks for the post. We will discuss your suggestion and post an update (sometime next month.)
Closed cespos closed 1 year ago
jmichel80
commented
2 years ago hi @cespos - thanks for the post. We will discuss your suggestion and post an update (sometime next month.)
 cespos
commented
2 years ago
cespos
commented
2 years ago Great, thanks!
 lohedges
commented
2 years ago
lohedges
commented
2 years ago Hi @cespos. Thanks for your query, this is really helpful. The leap_commands feature was added to solve a particular problem, i.e. the addition of custom parameters, and we obviously didn't think about all use cases, such as where you would need to inject extra commands in a different place within the LEaP script.
As you say, ideally it would be possible to pass a fully custom config file, or modify it as you can do for a Process. The issue is that some parameterisation functions have multi-step protocols, which call out to several backends. This means that there isn't a single configuration for the whole thing. In other cases, we might support both LEaP and pdb2gmx for the same FF, so the configuration options aren't universal.
I'll have a think at how to best implement this, then run some suggestions by you.
Cheers.
 AdeleHardie
commented
2 years ago
AdeleHardie
commented
2 years ago hi @cespos, you can also use the leap_commands parameter to almost have a custom config file. Here is an example for your case:
import BioSimSpace as BSS
pdb = BSS.IO.readMolecules('4LYT_Fixed.pdb')
# define custom leap script except for ff14SB sourcing
custom_commands = ['4lyt = loadPDB leap.pdb', # note the use of 'leap.pdb'
'bond 4lyt.6.SG 4lyt.127.SG',
'bond 4lyt.30.SG 4lyt.115.SG',
'bond 4lyt.64.SG 4lyt.80.SG',
'bond 4lyt.76.SG 4lyt.94.SG',
'saveamberparm 4lyt custom.prm7 custom.rst7']
parm = BSS.Parameters.ff14SB(pdb.getMolecule(0), leap_commands=custom_commands)
# load the custom molecule directly from working dir
parm_mol = BSS.IO.readMolecules(f'{parm.workDir()}/custom.*')
BSS.IO.saveMolecules('4LYT_parm', parm_mol, ['rst7', 'prm7'])I run the following to check that the S-S bonds are present in the topology:
$ cpptraj
> parm 4LYT_parm.prm7
> bondinfo :6@SG
# Mask [:6@SG] corresponds to 1 atoms.
#Bnd RK REQ Atom1 Atom2 A1 A2 T1 T2
1006 227.00 1.810 :6@CB :6@SG 96 99 2C S
1007 166.00 2.038 :6@SG :127@SG 99 1914 S Syou can see that SG in residue 6 is bonded to SG in residue 127
lohedges
commented
2 years ago We should easily be able to detect the present of disulphide bonds using Sire, then generate the correct LEaP bond command strings. For example, we could do something like following (using Sire 2023.0.0):
import BioSimSpace as BSS
from sire.legacy.Mol import Connectivity
from sire.legacy.Mol import Element
S = Element(S)
# Load the molecule.
mol = BSS.IO.readMolecules("4LYT_Fixed.pdb")[0]
# Generate the connectivity (bonding) object for the Sire molecule, then
# add this as a property.
connectivity = Connectivity(mol._sire_object)
mol._sire_object = mol._sire_object.edit().setProperty("connectivity", connectivity).molecule().commit()
# Get the bonds associated with the Sire molecule.
bonds = mol._sire_object.bonds()
# Create a list to store our disulphide bonds.
disulphides = []
# Loop over the bonds and store the disulphides.
for bond in bonds:
if bond.atom0().element() == S and bond.atom1().element() == S:
disulphides.append(bond)We then just need to loop over the disulphides list to create appropriately formatted bond records for LEaP. I'll see if I can do this when I get a chance.
lohedges
commented
2 years ago Okay, I've got this working on the feature-disulphide branch. The actual function that generates the records is here, which is basically the same as above with the addition of generating the record string for each bond.
The one thing that I'm concerned about is that determining the bonds via the Sire.Mol.Connectivity object relies on sane coordinates, i.e. it uses a bond hunter to determine bonds bases on equilibrium values for given elements. If you could share the 4LYT_Fixed.pdb file, then I could test that it generates the correct records in this case. (Any other appropriate file salong with the required bond records would be great too.)
lohedges
commented
2 years ago I had a quick Google and found a file with the same name in an AMBER tutorial here. Using this, I only generate two bond records, i.e.:
import BioSimSpace as BSS
m = BSS.IO.readMolecules("4LYT_Fixed.pdb")[0]
BSS.Parameters.Protocol._protocol._get_disulphide_bonds(m._sire_object)
['bond mol.6.SG mol.127.SG', 'bond mol.64.SG mol.80.SG']Perhaps this file is different, or maybe the Connectivity is not a reliable way to determine the bonds. (I'll see if there's a way of tuning the bond tolerance.)
lohedges
commented
2 years ago I can get it to work by tuning the tolerance, but since the bond hunter is based on covalent bonds, then I also need to tweak the maximum bond limit. I'll see what settings are most reliable. In practice, it might be best to add a new bond hunter specifically for disulphide bridges.
AdeleHardie
commented
2 years ago Could Sire parse CONECT or SSBOND records in a PDB when reading in a molecule? I think relying on the coordinates matching would make things very difficult if not impossible in some cases (e.g. mutating a residue to Cys and then not having the sane coordinates yet). LeAP itself uses CONECT (so if correct CONECT records are set, the ss bond setting is unnecessary), as an example CHARMM-GUI uses SSBOND. Here is my fixed file for 4LYT with those records included.
lohedges
commented
2 years ago Thanks, that is also an option. To date we haven't parsed those records since they are often missing or incomplete, so it's hard to detect when things are wrong. Since we rely on a well formatted PDB for paramterisation in general, I think it might be best to assume well formatted CONECT records too. This also gives the user an easy way to specify things precisely outside of BioSImSpace too. Note that we do have code that autogenerates a CONECT record based on the Connectivity, but obviously this is also reliant on sane coordinates.
lohedges
commented
2 years ago Ah, didn't realise that Sire also has a ChemicalBondHunter. This appears to work perfectly (for this PDB) with the default settings. Perhaps we could autodetect things if CONECT records aren't present, but use them when they are.
lohedges
commented
2 years ago I've made some edits to Sire to make the bond searching more flexible. The user can now specify the maximum search distance and a tolerance to use when comparing equilibrium bond radii for determining bonds. I've then tuned the parameters so that it works well for a few test systems.
I'll let some other collaborators test this against some of their input in order to work out where the edge cases are. It should be easy enough to fall back on the CONECT records, if present, or allow the user to specify things directly.
Cheers.
cespos
commented
2 years ago Hi @lohedges, Sorry I saw this only now. I will test this functionality asap and let you know how it works for me. Still feature-disulphide branch, right? Many thanks!
lohedges
commented
2 years ago Yes, that's the correct branch. The way in which I search for disulphides is quite flexible to allow for non-ideal configurations. It would be good to know if it's too flexible, e.g. picking up things that it shouldn't.
I'm also working on some improved CONECT writing ability. Currently we can generate these for all bonds, but I am tweaking to prune this to atoms non-standard residues and disulphides only. This means we might just be able to generate the CONECT records, rather than needing to add to the tLEaP input file. (Which approach is more robust, I'm not sure, although they use roughly the same underlying code.)
lohedges
commented
2 years ago Related to other discussions here and here, I've made some improvements to the Sire.IO.PDB2 parser to improve the handling of TER and HETATM records,which should hopefully make parameterisation more robust.
In doing so, I have switched to writing PDBv3 compliant CONECT records, which uses a similar approach to the one used to generate disulphide records noted above. We now write CONECT for all mandatory fields, i.e. disulphides and inter/intra residue bonds including any atom in a non-standard residue. (Obviously this approach requires sane PDB formatting, but this is a requirement for parameterisation anyway.) I've tested this and it works correctly for the input you provide, as well as some other examples that I sourced online.
One issue that I have discovered, which would also be a problem for the previous approach, is that the atom numbers used to generate the CONECT (or disulphide entries in the leap script) might not correspond the those in the PDB that is written, i.e. if a TER has been injected, or if the atom has been moved to a different location and renumbered. I am working on ways to resolve this, although I don't think it will be a common problem. I am guessing that we will need to write the PDB, load it back in, then generate the CONECT using the atom numbers from the file that was written (rather than from the original molecule).
Cheers.
lohedges
commented
2 years ago Oh, and I have now added a bonds option to appropriate parameterisation functions. This takes a list/tuple of atom pairs, which are then used to generate the LEaP bond directives. This suffers the same issue as above, i.e. the numbers in the molecule might not correspond to those in the PDB, so will need the same workaround, whatever that is.
lohedges
commented
2 years ago Okay, I've realised that I can match the atom coordinates from the Sire.System to those in the PDB file that is written in order to obtain the correct atom numbers to use for the CONECT records. This seems to work well and means that the CONECT will be valid regardless of what atom re-ordering occurs on write. Will test a little further then push an update.
cespos
commented
1 year ago Hi @lohedges, I have seen that the feature-disulphide branch was merged into devel. Today, I have installed the development branch and wanted to test the newly added feature. Could you please send me some instructions? Thanks!
lohedges
commented
1 year ago Assuming that you have a correctly formatted PDB file, the code should now autodetect disulphide bonds and add them to the LEaP script for you. This has been tested on a range of inputs and has been found to work well. If you find that it's not locating the bonds correctly, e.g. if the structure is a bit weird, then there a a few options:
1) Change the bond search tolerance or the maximum search distance. 2) Explicitly specify the atoms to bond by passing a list (or tuple) of atom pairs. (If any of these are found by the bond search, then they will be removed to avoid duplicates.)
See here for the documentation for ff14SB.
lohedges
commented
1 year ago There's also a unit test here that checks that the parameterisation works and that the resulting AMBER topology does contain the required disulphide bonds.
cespos
commented
1 year ago I tried but it did not work. Not sure whether I have the correct BioSimSpace version

lohedges
commented
1 year ago The current devel is version 2023.0.0+137.g63b14996 so you seem to be using an older version. (114 commits behind.)
cespos
commented
1 year ago Weird, I installed it today following the instructions. I'll figure it out and get back to you.
lohedges
commented
1 year ago It's possible that it's pulling in an older version because it doesn't want to change the version of Sire in your environment. To force it to use the latest you can do:
conda install -c conda-forge -c michellab/label/dev biosimspace sire=2023.0.2Or create an environment directly (which is recommended):
conda create -n bss -c conda-forge -c michellab/label/dev biosimspace sire=2023.0.2This worked:
conda install -c conda-forge -c michellab/label/dev biosimspace=2023.0.0=py39_137
cespos
commented
1 year ago The commands are now working... at least partially.

tleap however failed. The problem now is that tleap residues/atoms numbering should start from 1 and should be continuous. When I checked the leap.pdb in the temporary work_dir, I found that residues numbers were not continuous (no residue 16):

As a consequence, the bond specifications are not working. A similar issue will occur when proteins have multiple chains and there are disulfide bridges involving the different chains. The solution for those cases is to first run tleap to generate a processed pdb file (with continuous residues/atoms numbers). Then, check whether there are any disulfide bridges and generate the tleap bond commands. Finally, run tleap again to parametrize the protein with disulfide bonds. See also this.
lohedges
commented
1 year ago This is because your original input PDB file does not have continuous residue numbers. With BioSimSpace we make the assumption that the user input file is the source of truth and has been formatted appropriately. As such, we write back things like residue numbers as is, since the user might want to cross-reference things later down a pipeline. (We don't change things behind the users back.)
If this is a common issue with PDB input files, then could you provide some examples (particularly the multiple chain issue you mention). It might be possible to fix the files, i.e. make things continuous, then remap to the original numbering after the parameterisation is complete. (We already make use of a compatibility function that maps the output topology of tLEaP back into the one that was passed in, i.e. preserving naming etc.)
cespos
commented
1 year ago Hi @lohedges, Sorry again for taking a long time to follow up. This is the example of a protein that has disulphide bridges in two different chains, 3G8K. While this is the example of a protein having a bridge between two different chains, 5Y42.
I was thinking... Assuming that the user has not preprocessed the PDB file to have continuous residues numbers, could it be a solution to write the tleap bond lines extracting from the molecule sire_object the residue indices instead of the residue names?

lohedges
commented
1 year ago Thanks for this, I'll look into it next week.
lohedges
commented
1 year ago Assuming that the user has not preprocessed the PDB file to have continuous residues numbers, could it be a solution to write the tleap bond lines extracting from the molecule sire_object the residue indices instead of the residue names?
Sorry, perhaps I'm misunderstanding something here, the bonds do you mean use the residue indices instead of the number used in the original PDB, rather than the name? If LEaP just cares about the index, rather than cross-referencing it with the PDB, then this should be a very easy fix.
lohedges
commented
1 year ago I mean, the current bond record uses the residue number and name from the PDB, so just the name part should be replaced with the index?
cespos
commented
1 year ago Sorry if I was unclear. But yes it should be an easy fix :)
If I am not mistaken, LEAP just cares about indices when reading user specified bonds (indices starting from 1).
So, in the LEAP file in the disulfide bond definitions, instead of specifying residue numbers read from the PDB (e.g. bond mol.11.SG mol.72.SG), one could give the residues indices + 1 from the sire_object.residues, e.g bond mol.11.SG mol.71.SG.
To be honest, I did not test this out on the complicated cases that I sent you, but I can try to do that later before you make any changes!
Thanks!
lohedges
commented
1 year ago Great, that would be really helpful.
cespos
commented
1 year ago Hi Lester! I made a few tests.
If you use the function _get_disulphide_bonds() for 3G8K, you'll get this list of bonds:

Notice that the list has "duplicate" bond specifications. However, these are not really duplicates but are due to the presence of two chains with equal residue numbering.
I tried to parametrize this protein with tleap and confirmed that tleap only needs residue indices (in continuous order from 1) to specify disulfide bonds.
However, one still needs to generate a temporary PDB file (even within tleap) with the "correct" residue numbering.
Here is the tleap configuration file that I used:
source leaprc.protein.ff14SB
source leaprc.water.tip3p
mol2 = loadPdb 3g8k_moe_prep.pdb
savepdb mol2 3g8k_new.pdb
mol = loadPdb 3g8k_new.pdb
bond mol.10.SG mol.15.SG
bond mol.28.SG mol.116.SG
bond mol.32.SG mol.118.SG
bond mol.97.SG mol.110.SG
bond mol.139.SG mol.144.SG
bond mol.157.SG mol.245.SG
bond mol.161.SG mol.247.SG
bond mol.226.SG mol.239.SG
saveAmberParm mol 3g8k.top 3g8k.crd
quitNotice also that using the indices, there are no duplicate bond definitions.
So... it should be an easy fix to the _get_disulphide_bonds() function with the addition of two lines to the tleap configuration file.
I tested also the 5Y42 case and worked as well.
I am attaching here prepared PDB files and tleap configuration files (with txt extension). Please, let me know if I can do anything else to help.
Best, Carmen
lohedges
commented
1 year ago Thanks for this, I'll create a new feature branch and see if I can get it working. For the purposes of writing the file for LEaP it should be okay to manually re-number the residues, since we will keep the originals when the force field parameters are copied back from the topology file that is generated.
lohedges
commented
1 year ago This is now implemented here. Note that using your files directly, I can only get the 3G8K one to work , and only when using ff14SB. With the other file, LEaP complains about missing atom types, and using force fields other than ff14SB with 3G8K causes LEaP to add missing atoms, which breaks our input/output compatibility checks for the parameterisation.
cespos
commented
1 year ago Hi! It looks like it worked!! All my examples run through! I think we can close this issue and merge to main branch. Many thanks @lohedges!
lohedges
commented
1 year ago Fantastic. Thanks for confirming. I have another external collaborator who is also testing the update against their benchmark set. I'll close this when they are happy that things are still working.
Thanks again for your input. It turns out that the solution was much simpler than I originally thought.
cespos
commented
1 year ago Thanks to you!
Is your feature request related to a problem? Please describe. I have tried to prepare a system containing 4 disulfide bridges, however SS-bridge parameters were not in the output parameter file. When generating the parameter file using tleap, it is necessary to specify the SS-bridges explicitly in the following format after the pdb has been loaded.
With BSS, it is not possible to insert these lines in the leap configuration file because although there is a leap_commands option, extra lines are written before the system is loaded.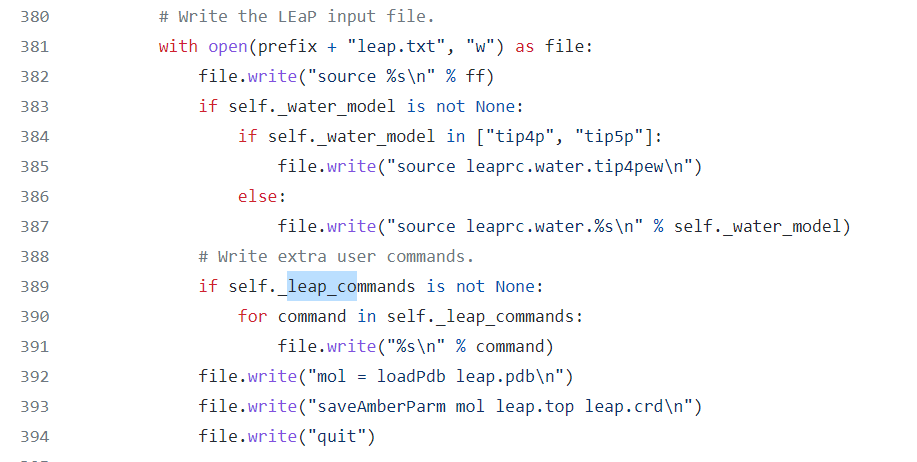
Describe the solution you'd like Would it be possible to have a leap_insert_flag, to decide where to insert the additional "leap_commands"? Something like "head", "pre-load", "post-load", "tail"?
Describe alternatives you've considered Or alternatively, would it be possible to have the option to specify your own tleap configuration file (as it is possible for MD configuration files)? ...Maybe this alternative would be even more useful because will allow for more flexibility.
Additional context Add any other context or screenshots about the feature request here.