 mozack
commented
6 years ago
mozack
commented
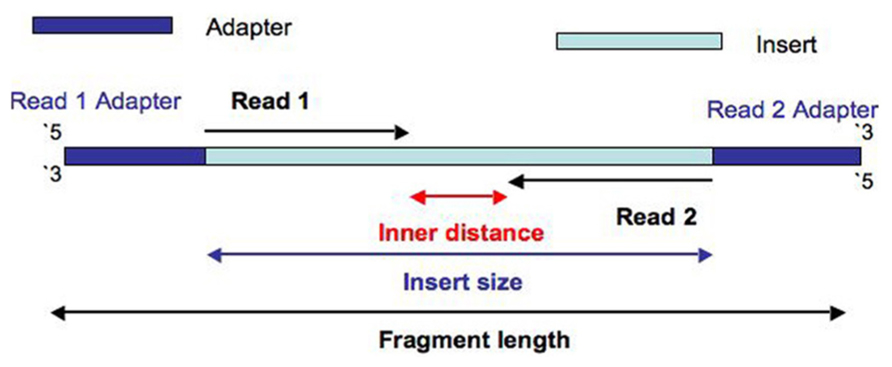
6 years ago This is the median insert size for the sample. See: https://www.frontiersin.org/files/Articles/77572/fgene-05-00005-HTML/image_m/fgene-05-00005-g001.jpg
{kind=link}
If the provider of the data you are using knows roughly the insert size, then you can use that. If not, you can map a subset of your reads to a transcriptome reference using a non-splice aware aligner such as bwa mem.
For example, the following maps the first 1 million reads of a gzipped fastq pair to the $TRANSCRIPTOME reference.
bwa mem -t 8 -S $TRANSCRIPTOME '<zcat *_1.fastq.gz | head -4000000' '<zcat *_2.fastq.gz | head -4000000' 2> bwa.head.log | samtools view -1 -bS -F 0xC -f 0x02 - > bwa.head.bam
You can then use the TLEN value (column 9) from the resultant bam file to compute the median insert size.
Hi, I am not sure how to set the ins parameter and what does it mean, could you please help on this?