 BokaiZhu
commented
2 years ago
BokaiZhu
commented
2 years ago Closed StiThor closed 8 months ago
BokaiZhu
commented
2 years ago  StiThor
commented
2 years ago
StiThor
commented
2 years ago Hi again. I seem to have trouble finding the script for generating a similar file as the one mentioned above with my own data. I have IMC data and I run the Steinbock segmentation (Deepcell/Mesmer) from Bodenmiller to make the cell mask. However, this approach does not generate the necessary file needed to do the REDSEA. Could you link that particular part of the script in Deepcell needed to make the file?
BokaiZhu
commented
2 years ago Hi there I see the question here. The short answer is the method is not yet compatible with the segmentation you are using here. The Redsea method we described in the manuscript works on an earlier version of Deepcell (described ref1 and ref2): where Deepcell will produce a nuclear probability map instead of directly a whole cell seg map. We then use the nuclear prob map to run a watershed algo (here), and generate boundary pixels like this (where Redsea runs on the red pixels)

While the current segmentation you are using will directly produce a label map, and the potential boundary map would look like something:

As you can tell, they look slightly different and might cause some problems for the code, partially discussed here.
We do have a working script that is compatible with these newer versions of segmentation map, but we do need some time before we can be confident enough that is safe to use before we release it. For now, I will make this issue open and happy to discuss further.
StiThor
commented
2 years ago I see :) Very exciting that you guys are working with something that works with the newer version of Deepcell since our experience is that this segmentation works quite well! Have you thought of trying to have the REDSEA built in the code for Steinbock Deepcell so the whole process could be automated? If you need some Guiney pigs to test these new scripts on that are compatible with the newer version of deepcell I`m your guy :)
 janinemelsen
commented
1 year ago
janinemelsen
commented
1 year ago Hi! I was wondering whether there is any update on using REDSEA in combination with the current mesmer segmentation:)
 yunhaoBai
commented
1 year ago
yunhaoBai
commented
1 year ago Hi there, I wrote some new version which is compatible with the Mesmer segmentation, we have validated that it can give results, but never have the chance to re-validate the scripts again with all the extensive experiments we did in the original paper. REDSEA_Mesmer.zip You may check the attached files
janinemelsen
commented
1 year ago Thank you so much! I am bit confused on my input files.
I have a whole cell segmentation mask generated by mesmer as included in the mcmicro pipeline (see also discussion here: https://forum.image.sc/t/nuclei-probability-matrix-from-mesmer-using-mcmicro/62720)
janinemelsen
commented
1 year ago @yunhaoBai could you please help me further in the usage of your new version?
 BenCaiello
commented
8 months ago
BenCaiello
commented
8 months ago Hello! I recently starting looking at how I might consider incorporating your helpful package into my IMC workflows -- but like others I have been using steinbock to produce images and segment, so the file & mask formatting is all wrong.
I have attempted to edit the python jupyter notebook file currently in the note into a new form that can take up steinbock output (multi-channel .tiff image and DeepCell masks without 0 boundaries). I don't have matlab, and am also much more familiar with python so I started there.
It looks like the modified script might be working (although it is slower because of the lack of easily identified boundaries between cells requiring the boundary identifying step to iterate over a lot more of the image), but what would be the best way to me to confirm that the compensation is occurring as intended? Normally, I might plan to try running the example data on my script, but of course that won't work because of the differences in the mask format.
If you want to see my edited script, I'd of course be happy to share it with y'all!
BokaiZhu
commented
8 months ago Hi BenCaiello! Thanks for modifying the script so that it can take Steinbock outputs. I have not used this method before and am unfamiliar with it, so it is always good someone with the expertise can rewrite our code! Also I'm happy the python code was helpful, considering it was wrapped up in a very short time frame.
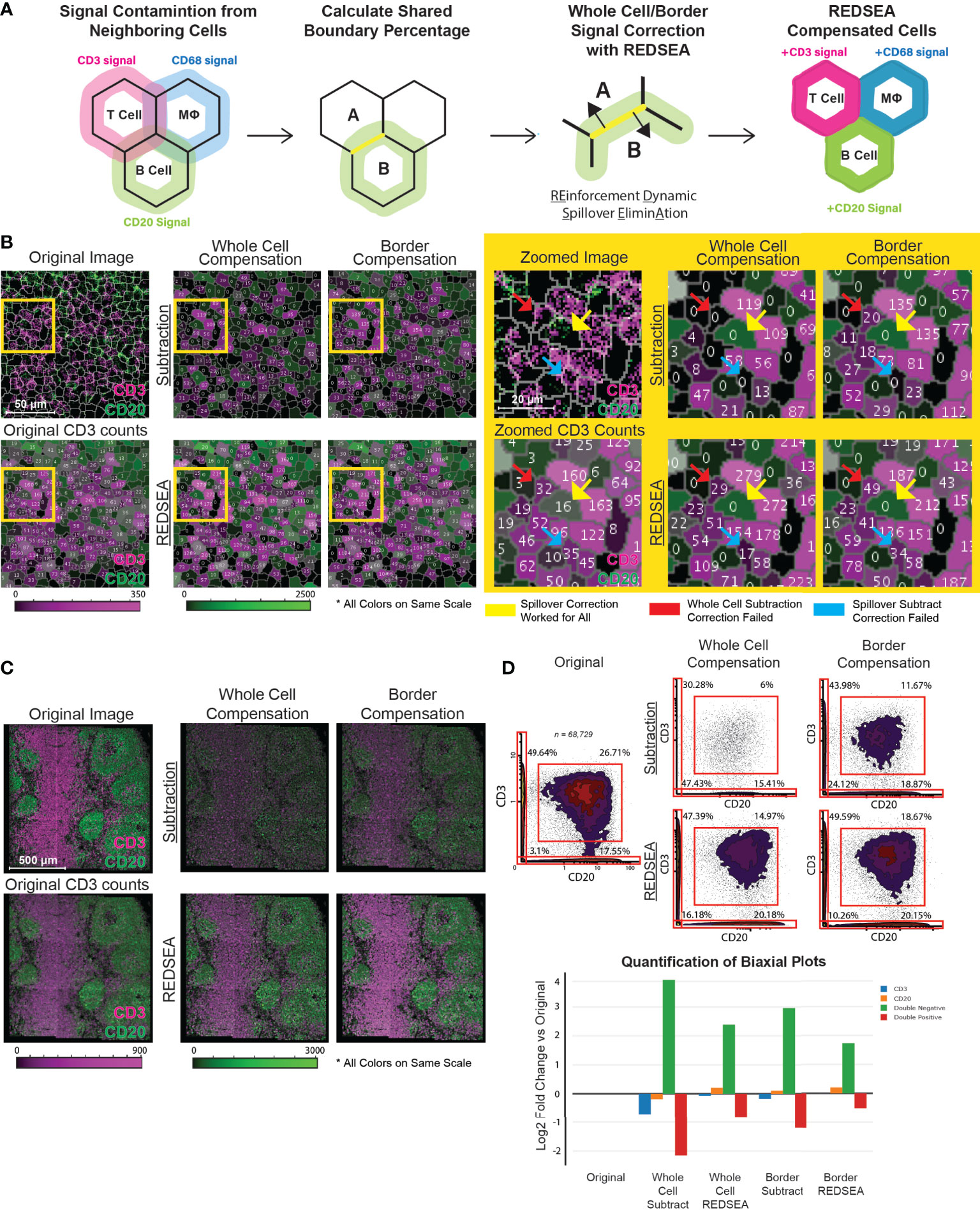
In terms of benchmarking, perhaps I would suggest similar to what we have done in the manuscript figure 1D, where if method working you should see eg CD3-CD20 double positive cells get lower. I understand this would not be a perfect benchmark, instead of replicating all the analyses we did in the original manuscript (which would be too much work I guess).
Happy to discuss more if need input on this! Apologies in advance if slow response, both myself and @yunhaoBai graduated from the Nolan lab last year so we were not checking-in this repo as frequently as before.
Best, Bokai
BenCaiello
commented
8 months ago I finished updating my version of the script and I did run data through it and got results that look reasonably in line with figure 1D:
y-axis (parameter 16) is CD20, x-axis is CD3, both arcsinh transformed. Plotted in FlowJo, the RedSEA compensation is on the right. Data from this public dataset of IMC images of tonsil tissue: https://zenodo.org/records/8023452
Are there other tests that I should consider doing?
I opened a fork of this repo & have my script inside it in the REDSEApy folder if you want to see it.
Thanks so much for your input! Ben
StiThor
commented
8 months ago Hi BenCaiello! Very nice that you have taken the time to make the RedSea for steinbock. I have around 400 tissue samples (2.2 mill cells) and will try to run your script. When in the pipeline do you recommend to do IMC Denoise?
BenCaiello
commented
8 months ago Hello StiThor, I hope the script works well for you! I have not tested it thoroughly & am somewhat new to all this so let me know how it works or if there are any errors you encounter! I want to check that it is implementing the algorithm correctly, and I may also update the script at some point to allow it to ingest whole folders of image data at once -- right now it is only set up to do 1 image at a time, with the user needing to input the new file name each time it is run.
For IMC_denoise -- you noticed that I also have tried setting up a python script for IMC_denoise + steinbock integration? So far I have mostly denoised my segmentation markers to improve the output of DeepCell, since the cytoplasmic markers in the datasets I've seen are often rather poor. So, in my case, I denoise immediately before segmentation with steinbock.
Again, though, I am pretty new to this (both python scripting & IMC data analysis) and have been experimenting with these packages because I'm helping with the set-up of a new, more standardized bioinformatic pipeline for IMC / MIBI data at my institution -- so I suspect you have a much deeper experience with IMC_denoise and REDSEA, and I would love to know what your experience is with them / what you've been using them for!
BenCaiello
commented
8 months ago I have kept working on the script and now have a second version in my fork that takes in entire folders of images at once and is better integrated with steinbock (does not require an extra massDS.csv file, instead it now use the native steinbock panel.csv file).
However, I have also definitely identified some errors in the script's algorithm, so I'd recommend not using it until I can fix those!
BenCaiello
commented
8 months ago I think I've completed my version of the steinbock / python integration script, and am fairly satisfied that the algorithm is working as intended! The script is in my fork of this repo in the python folder under the label "REDSEA_for steinbock_DeepCell_BPC_multi_file_final.ipynb". If you try it out, I'd be interested in seeing how it goes.
It should be able to process many images at once and be well integrated with the native output of steinbock -- although it looks like the size / shape parameter is very important to tune. With the practice data I had been using, the cells were very small and the "border" regions often comprised the majority of the cell.
One other thing I've noticed though is that this repo has no license attached to it -- so technically it is not legal to use right now? Presumably just an oversight given what the authors have said, so I'll open a separate issue for that question.
BokaiZhu
commented
8 months ago Congrats and thanks for writing up the updated script! This would be really helpful for the community! I will definitely point people into this direction if they want to use redsea in the context of steinbock!
For license I will fix in the separate ticket.
Best, Bokai
{kind=link}
How was the MIBI feature_1_frame_1_p1_dsDNA file generated?