 Yoshanuikabundi
commented
2 years ago
Yoshanuikabundi
commented
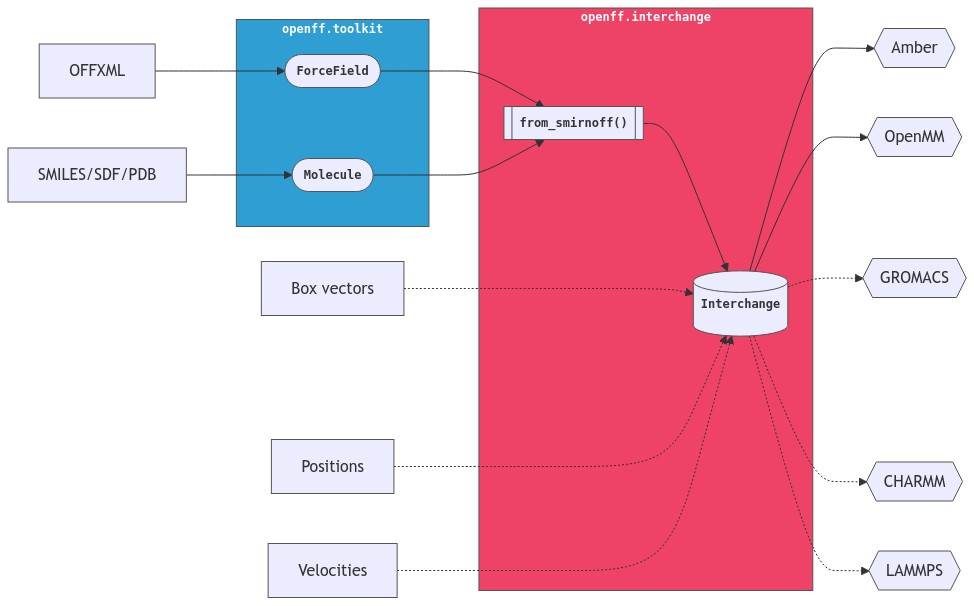
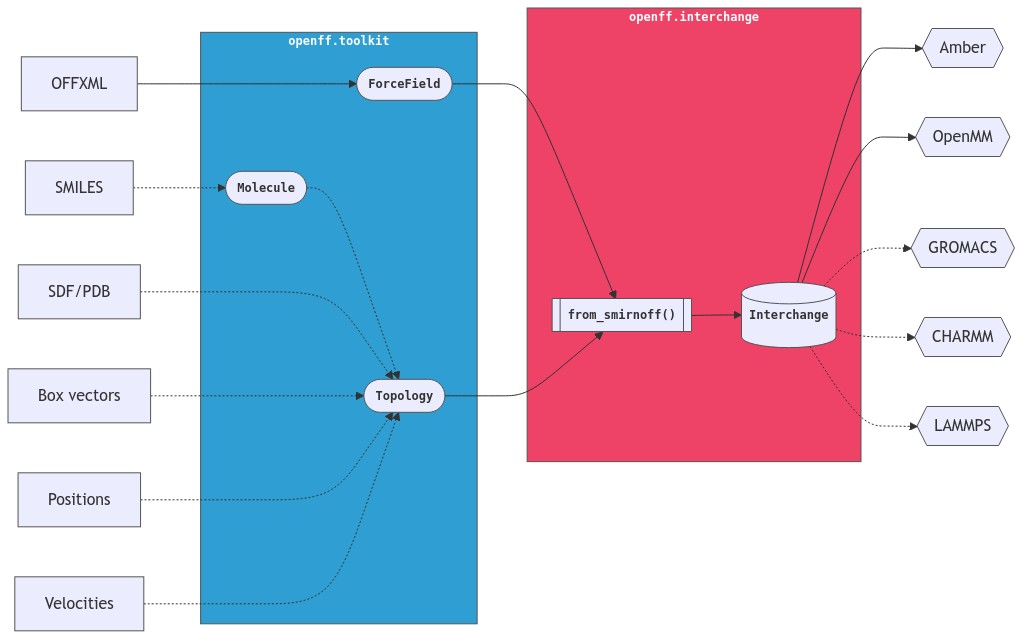
2 years ago This refactor would also make it really clear what arguments belong on the parameterize methods. At the moment, box is an argument to from_smirnoff, but positions isn't, despite both pieces of information potentially being present in the Topology.
By breaking the method up, users can easily understand that a from_topology method takes all the information from the topology, and if they want to override something they need to use attribute assignment. No-one feels awkward about sometimes being able to do everything in one method call and sometimes not. There's a clear separation of concerns between topology information and force field information.
 mattwthompson
mattwthompson

 SimonBoothroyd
SimonBoothroyd j-wags
j-wags
This is part of my crusade against API consumption of
Topology:stuck_out_tongue:Would it be crazy to refactor the parts of
from_smirnoffthat apply the force field out into anparametrize_smirnoffmethod that everyone can use? I'm imagining it would open up possibilities of adding morefrommethods that include topology information but not a forcefield, and then applying the force field in a separate step. For example:Alongside
Alongside
And
And
This would mean users could prepare their system however they like, bring the system into Interchange with the appropriate method, and then parameterize it.
An alternative is to keep things how they are now, where these sorts of construction methods are implemented on
Topologyandfrom_smirnoffplays the role of parameterizing. This means users have to keepTopologyin their mental model.An alternative method name would be
add_smirnoff,add_foyeretc.As a side note, does
from_smirnoffstore the entire force field in the Interchange or just the potential handlers that are used in the topology? If it stores the entire force field, you could also provide similar capabilities in the form ofadd_pdb,add_mdtraj,add_moleculeetc and make thetopologyargument offrom_smirnoffoptional.