 oushujun
commented
3 years ago
oushujun
commented
3 years ago Hi Weihan,
Thank you for your detailed descriptions. I open a new issue because this is a different but interesting topic.
We demonstrated in the LAI paper, that genome size, TE content, contig N50, BUSCO, etc, are not significantly correlated with LAI. Of course back then we only had a limited number of 'good' genomes to test on, and their quality was not evenly distributed. I have noticed similar cases but those are sequenced by Nanopore. I thought it may have something to do with the sequencing technique, but from your case it may be more prevalent. I am still collecting similar cases because so far it is sporadic and thus I have limited power to detect the cause. By the time I got enough data, the raw LAI correction may need a re-calibration.
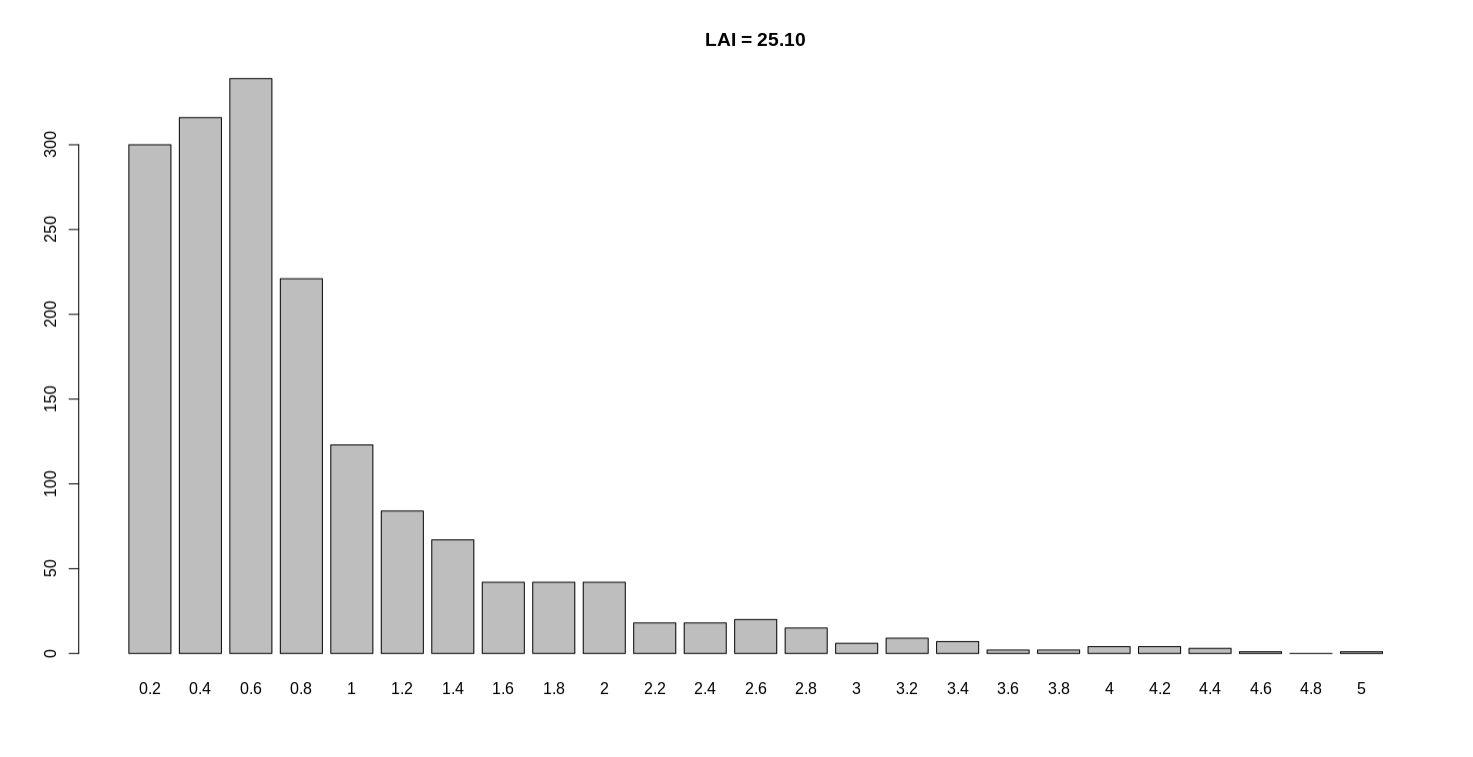
You mentioned assembling the genome with >20kb subreads, what is the coverage? Can you post one of the LAI screen outputs for this genome?
Thanks, Shujun
 Weihankk
Weihankk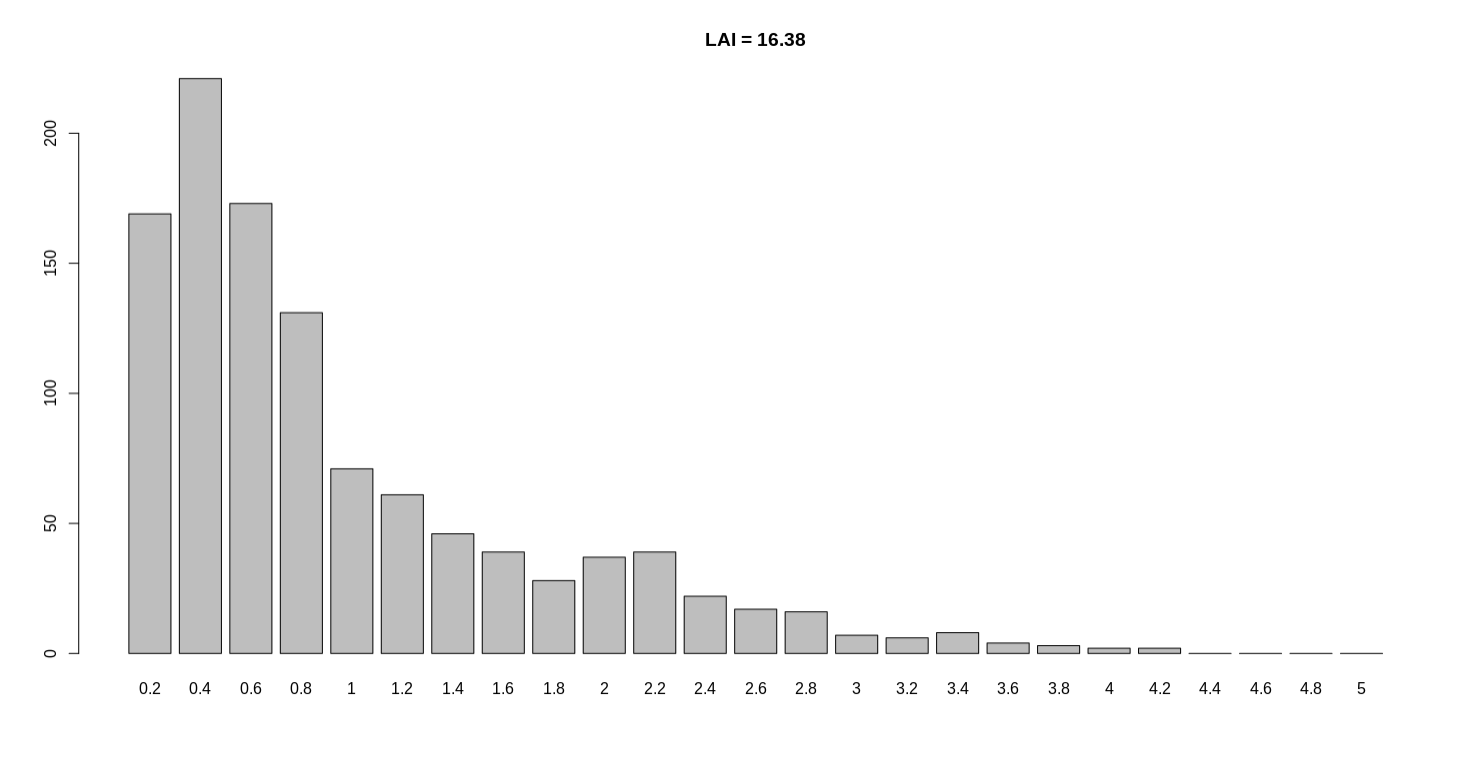
 chaimol
chaimol frabanal
frabanal
Dear shujun, First of all apologize for my bad English. I have sequenced dozens species, these species belong to the same genus (eg. wild, cultivars, landrances), and I will construct a pan-genome. Sorry I am not an expert in sequencing experiments either. But I know they all use the same kit and extraction method to build the library.
Here I give you a detailed description of the only sample with LAI < 20. This sample named SampleA. At the beginning of my project, dozens of samples were all sequenced about 120 ~ 160X by Pacbio. The subreads length also well and subreads N50 are 10 ~ 20k. This looks really good so I assembled them by CANU and obtained dozens of genomes. The LAI value of all these genomes exceed 20 except SampleA is 16. And the contig N50 of SampleA also very unusual (just 200kb). So we contacted technical experts to perform another round sequencing. This time I got about 500X new Pacbio data of SampleA, also with normal subreads length and subreads N50. I assembled SampleA and run LTR_retriever, the contig N50 improved to ~5Mb while the LAI is still ~16. Surprised me and incomprehensible. Since I have enough data, I also tried only use length > 8kb, 10kb, even 20kb subreads to run different assembly software, all the LAI values is between 16 and 17, stable as Mount Tai. According to our common sense and your articles published on Nature communication, high-depth sequencing will improve assembly quality. The contig N50 is significantly improved (200kb to 5Mb), but the LAI value no improvement.
While writing here, I thought about it again. If it is a problem with library building, the contig N50 will not be improved. I used to think there was a problem with the DNA extraction process, human factors like some experimental operations. However, I got reasonable geome size, high contig N50 and high BUSCO (99%). So my guess last time about sequencing library construction may be unreasonable. The problem that still bother me is high-depth sequencing assembly get a long contig N50 but low LAI value. Different assembly methods and parameters have an effect on contig N50, but it doesn’t seem to have effect on LAI. Even I only use subreads length > 10kb to run assembly the LAI is still ~16. I have high-depth sequencing and long reads, the LAI hasn’t improved.
Thank you for taking the time to discuss so much with me.
Best regards, Weihan
_Originally posted by @Weihankk in https://github.com/oushujun/LTR_retriever/issues/86#issuecomment-768498135_