 PauBadiaM
commented
7 months ago
PauBadiaM
commented
7 months ago Hi @Gregjlt,
To compute enrichment scores we use the univariate linear model method:
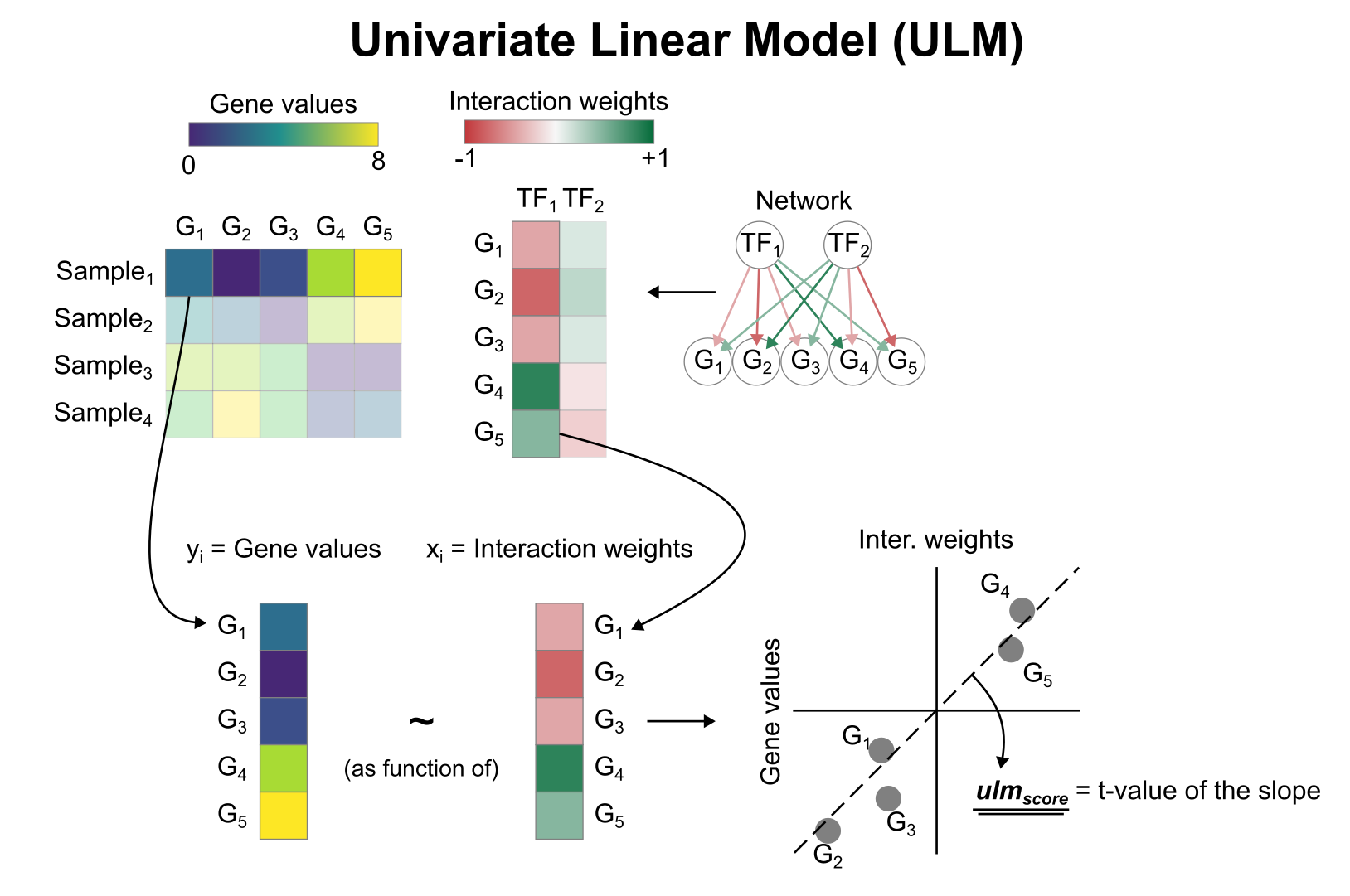 In this model where each gene is an observation, the response variable (y) is the change in gene expression and the explanatory variable (x) is the weight for that TF-Gene interaction. In the case of SP1, the red dots that have positive t-values belong to the first quadrant (+ and +), and the blue dots that have negative values belong to the 3rd quadrant (- and -). If you fit a line (like the one shown in the attached image) you would get a positive slope. The moment you have the contrary, blue dots with positive t-values (- and +) and/or red dots with negative t-values (+ and -) the slope becomes negative.
In this model where each gene is an observation, the response variable (y) is the change in gene expression and the explanatory variable (x) is the weight for that TF-Gene interaction. In the case of SP1, the red dots that have positive t-values belong to the first quadrant (+ and +), and the blue dots that have negative values belong to the 3rd quadrant (- and -). If you fit a line (like the one shown in the attached image) you would get a positive slope. The moment you have the contrary, blue dots with positive t-values (- and +) and/or red dots with negative t-values (+ and -) the slope becomes negative.
Basically, you can get a positive activity by having most positive targets with positive t-values, or by having most negative targets with negative t-values, or a combination of both. The moment this becomes inconsistent, for example, a random mixture of positive and negative targets with either positive or negative t-values, there will be no trend and the slope of the linear model will be flat.
Hope this is helpful! Let me know if you need any further clarification.
 Gregjlt
Gregjlt
Hello, I'm having trouble understanding the interpretation of your example for the transcription factor activity inference in bulk RNA-seq, particularly on this graph :
Looking at the code below, genes in red, are the ones that have the same sign for the MoR and the t value, and in blue the ones that have opposite signs. So, for the best predicted TFs, I would expect to have the majority of genes that are represented in red, as this means that the logFC observed corresponds to the type of regulation predicted by the MoR. However, we can see that for SP1, one of the best predicted TFs, almost half of the genes are in blue, so have a t value in opposite sign with the MoR. Am I understanding something wrong in this example ?
Thanks for your answer !