 PauBadiaM
commented
4 months ago
PauBadiaM
commented
4 months ago Hi @ngvananh2508,
Indeed decoupler can achieve this. There are two ways on how to do it:
- Compute pathway activities at the single-cell level and then summarize per cell type as shown in this vignette
- Generate pseudobulk profiles, perform DEA and then compute activities, as shown in this other vignette.
Hope this is helpful!
 ngvananh2508
ngvananh2508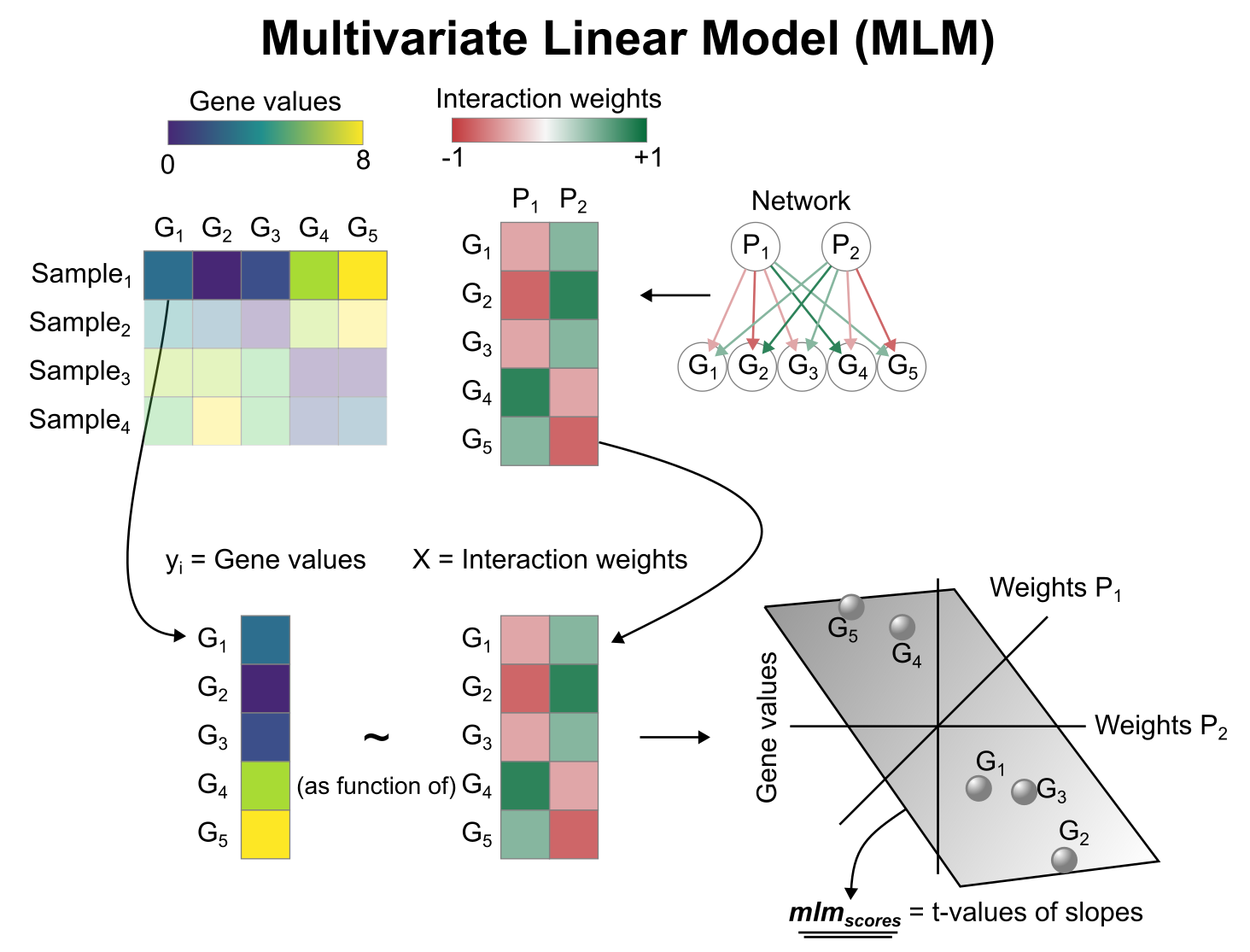
Describe your question Does your library have the method can detect which signaling pathway is down or up-regulated and does it have method to define signaling pathways for each cell type instead of each cell? I saw that you library have ora and aucell methods can detect the enriched signaling pathways that appear in each cell.
Thank you very much.