 grst
commented
4 years ago
grst
commented
4 years ago Hi @jfx319,
thanks for your extensive feedback, we appreciate it a lot!
A) public vs. private
I agree that it's better to stick with the historical meaning of "private" and "public" clonotypes and come up with a new terminology for clonotypes shared between tissues of the same patient.
In the publication of Wu et al. they use "dual-expanded" for clonotypes that are expanded in both blood and tumor sections. I'm not a big fan of that term as it is easily confused with "dual TCRs" having more than a single pair of alpha- and beta chains.
What do you think of tissue-specific vs multi-tissue/cross-tissue clonotypes? Or tissue-private vs. tissue-public?
We thought of augmenting our docs with a glossary (#108), this would be the perfect place to add the definitions once we agreed on a term.
B) definition of a clone
We initially considered including the CDR1 & 2 regions in our model, but found that, unfortunately, it is not straightforward to retrieve the corresponding information from cellranger or TraCeR. Therefore, we decided to stick with CDR3-only for the beginning, as this is what people are most interested at the moment.
That being said, we still believe that it would be a good idea to include this information and will re-consider it later on. For TraCeR, the CDR1 and 2 regions are indeed available, but hidden in some secondary output files and require considerable parsing effort. Cellranger, to my knowledge, doesn't report CDR1 and 2 at all, but provides the entire contig, from which they could potentially be extracted.
The better solution, however, IMO, would be if the preprocessing tools were improved to make this information more readily available.
option to use UMI counts as opposed to read counts when calling the most abundant chain
We currently use UMI counts for this already, if available. I will clarify this in the docs.
C) additional way to distinguish observed chain categories from true categories
These are really good ideas, but they require considerable effort both for implementing and validating. Therefore, I created a separate issue (https://github.com/icbi-lab/scirpy/issues/116) for further discussion.
D) distance metric guidance
This is a good point, and we definitely should add a case-study on this. I can think of two approaches for validating distance-cutoffs:
- specificity assays based on Dextamer staining. There's a dataset available provided by 10x (-> Application Note - A new way of Exploring Immunity).
- For the BLOSUM62 approach, we could check in conservation data of various binding domains how much sequence-variance tends to be typically tolerated there.
In this context also @davidsebfischer's tcellmatch is interesting. He developed an additional, machine-learning-based metric for assessing TCR-specificity and also put quite some effort into validation.
Lastly, the case study link needs to be updated to the icbi-lab.github.io
Good catch, I updated the README.
@grst What a great tool, very much needed and appreciated! Thank you.
Towards making this better still and more enduring (including for VDJ biologists), I have a few comments/suggestions:
A) "public" vs "private" clone
Historically, T-cell biologists use these terms for comparing between 2 person's repertoire (see Turner et al 2006 review), not between tissues of the same person (as referred to in your tutorial). Since these terms have a lot of history, it's worth weighing how people will refer to it to minimize confusion. Perhaps, it's an opportunity to revisit the choice of words, and maybe come up with a better naming scheme that provides backwards-compatibility, yet offers new ways of distinguishing these nuances.
B) definition of a clone
A frequent assumption for defining a clone is by CDR3 region alone, as it is the most diverse due to bridging the junctional diversity and therefore, likely to be unique to a "clone". Naively, one would think that a repeat CDR3 sequence is exceedingly rare, but the NGS-based approaches have opened the door for empirically assessing these repeat occurrences -- and it seems that they do occur more frequently than one might think. In addition, more recent interest in rarer innate-like TCRs (iNKT, GEM, MAIT) have refocused some attention on the TCR binding contributions from CDR1 and CDR2 (entirely determined by the V region). Collectively, this might point towards a joint consideration of these multi-part contributions: {V and CDR3}, {CDR1, CDR2, CDR3}, or {entire TCR contig} as more precise ways of considering a "clone", including when thinking about convergent selection. As technologies continue to improve and make possible the measurement of these additional "features", they could grow in importance for a holistic model.
See Figure 1 of Attaf et al 2015.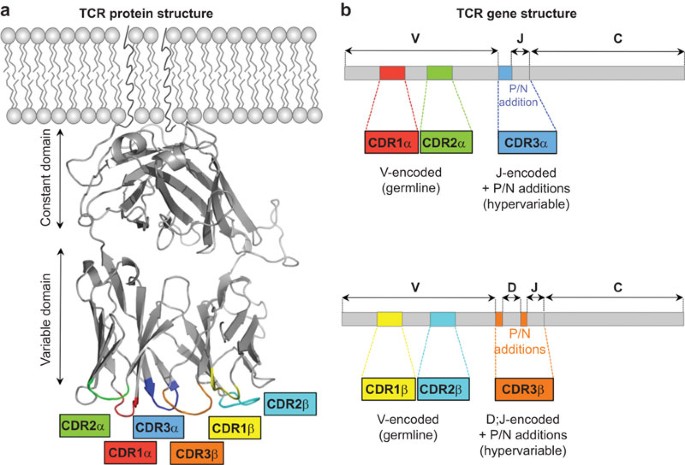
The utility of these other complementarity-determining regions becomes easier to appreciate in Kronenberg & Zajonc 2013 review's illustration of how TCRs can vary their binding: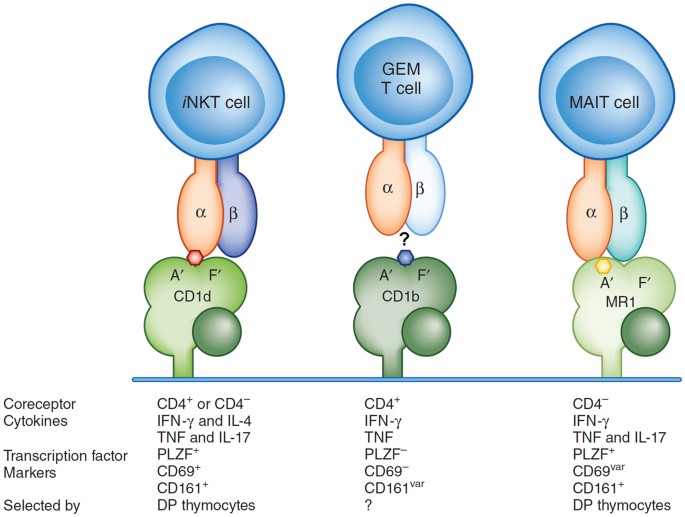
Single-cell specific considerations:
C) additional way to distinguish observed chain categories from true categories
At least several factors affect observed categories:
Adding to the sources you've already cited, this illustration from Dupic et al 2019 emphasizes the different a priori expectation of the beta vs alpha chain:
Separate from the allelic exclusion bias, this figure from Redmond et al 2016 illustrates the 2 fold expression bias between alpha and beta: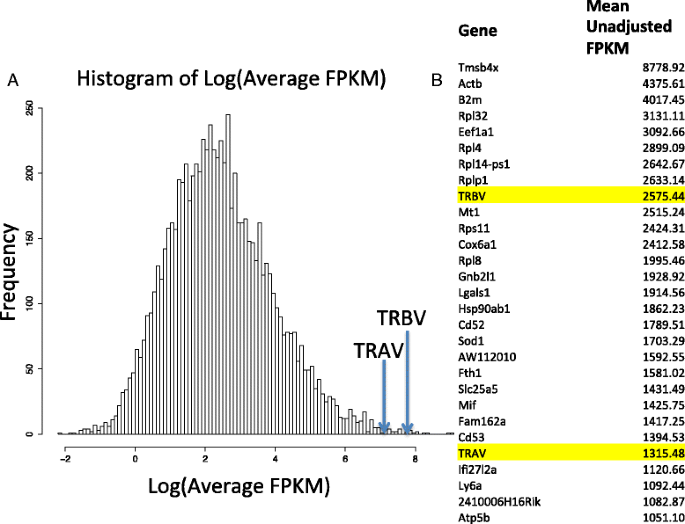
These biological mechanisms contribute the the observed frequencies. But what is measured is additionally confounded by signal dropout and by doublets.
Single-cell considerations:
D) distance metric guidance
It's great that at least 3 metrics are provided. As the choice of distance metric as well as the thresholds used are not immediately obvious, it would be useful for biologists to have some guidance on choice of cutoff/thresholds to use, and their reasons/impact based on some justifiable empirical case studies). I like that a biologically-based BLOSUM62 matrix is one option (time will tell whether this or the levenstein metric ultimately applies better in this particular context, or maybe a custom metric based on all observed TCR sequences to date). For now, it's helpful to include some recommendations, what that effectively translates into, and why that choice is recommended based on your own case studies.
Per the same considerations for the definition of a clone, perhaps an additional parameter/method can eventually be implemented to take into consideration these additional specificity-determining features (e.g. CDR1, CDR2) when building the neighborhhood graph.
Lastly, the case study link needs to be updated to the icbi-lab.github.io