 marehr
commented
4 years ago
marehr
commented
4 years ago Hello @cartoonist!
We unfortunately haven't implemented a multiple sequence alignment, yet. But, we already had some discussions how to implement it in seqan3 and we definitely want to implement this feature (we also have some projects that need them). Currently, we are focusing on making the features that seqan3 already offers stable, e.g. the search and pairwise alignment.
Since you already have experience with seqan2, maybe you could help us design this feature by answering these questions:
- What use-case do you have and how did you implement it with seqan2? I think we had several ways how to use the MSA so it would be interesting to see what you need!
- Do you have publicly available code that we can look at? Or maybe you can provide some snippets?
- How did you call the MSA algorithm and was there something that you thought was exceedingly hard to understand, or unnecessary verbose, or hard to use (code-wise with regards to user friendliness)?
- How do you think should the interface look like? Maybe you have an idea of a slick and easy interface. Maybe you already peaked into seqan3 and have an idea how you expect it to look like. Maybe some expectation from other libraries or programming languages, like bio-python?
Thank you!
 cartoonist
cartoonist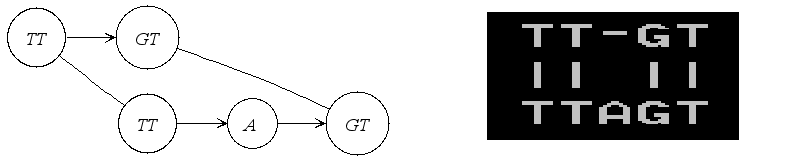
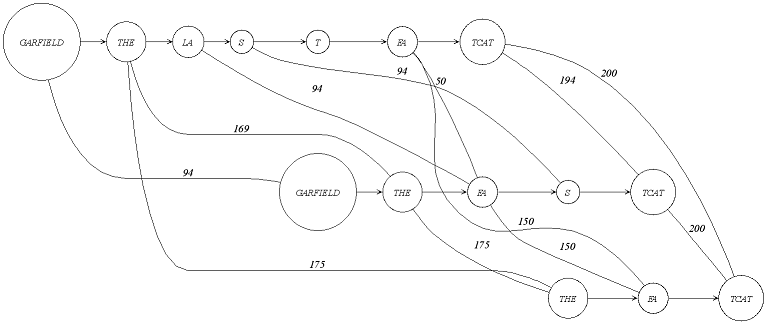 The title says: An alignment graph with 3 sequences.
The title says: An alignment graph with 3 sequences. rrahn
rrahn rlorigro
rlorigro smehringer
smehringer
Question
I have just started using seqan3. I have worked with seqan2 though. I want to ask about multiple sequence alignment and alignment graphs in seqan3. I could not find anything about MSA in the documentation. Are they missing in the documentation or they are not implemented yet?