 ximik69
commented
7 months ago
ximik69
commented
7 months ago My settings are - SG15 pp, 50Ha cutoff, 1E-8Ha energy diff threshold for elec-minimize,
1E-7Ha energy diff threshold for ionic-minimize, knormThreshold 1e-5. Thus the structures are converged quite well.
I optimize only atomic positions and parameters of the unit cell are fixed.
I use
coulomb-interaction Slab 001
coulomb-truncation-embed 0 0 0.5in order to mitigate interactions between slabs. But I see teeth on the surface energy vs. slab thickness plot.
 shankar1729
shankar1729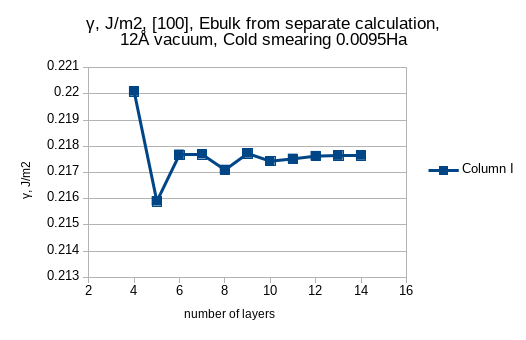




{kind=link}
Hello dear Shankar, I have encountered small teeth about 0.01 J/m2 while trying to find convergence of surface energy of Na {100} as a function of slab thickness. For me it is quite ok, because surface energy of Na {100} is 0.22J/m2 and I reproduce it. But how can I get rid of these teeth?
My workflow was the following:
The convergence in case 4a is not visible, in case 4b there is some tendency towards convergence and in case 4c no convergence is seen. In case 4a some linear slope is due to small difference between Ebulk from separate calculation and Ebulk from consecutive increase of slab thickness. This is mitigated in case 4b. I have deliberately chosen 8 layers as some tradeoff between accuracy and speed.