 sjteresi
commented
5 months ago
sjteresi
commented
5 months ago Hi,
Thank you for the helpful description. Please try moving the tmp directory elsewhere, as the function looks like it is still detecting the overlap files. Essentially, please make the folder that you are providing a terminal folder that only has the .h5 output.
If this is true, I will edit the error message to be more descriptive. Seems like having the .h5 files on their own in a directory is not enough, it cannot have the overlap files in a subdirectory.
Thank you for the tool you developed. I am trying to study the relationship between genes and TEs, which has helped me a lot, but I still know very little and need your help.
I have successfully run the example.
python process_genome.py **$DATA_DIR/Cleaned_TAIR10_GFF3_genes_main_chromosomes.tsv** $DATA_DIR/Cleaned_TAIR10_chr_main_chromosomes.fas.mod.EDTA.TEanno.tsv test -c config/production_run_config.ini -n 60 -o test333and get the result
I saw the script running in the readme.
python examples/general_read_density_data.py CLEANED_GENE_ANNOTATION.tsv DENSITY_DATA_FOLDER "Arabidopsis_(.*?).h5"I run like this
python examples/general_read_density_data.py $DATA_DIR/Cleaned_TAIR10_GFF3_genes_main_chromosomes.tsv test333 "test_(.*?).h5"
The error occurred.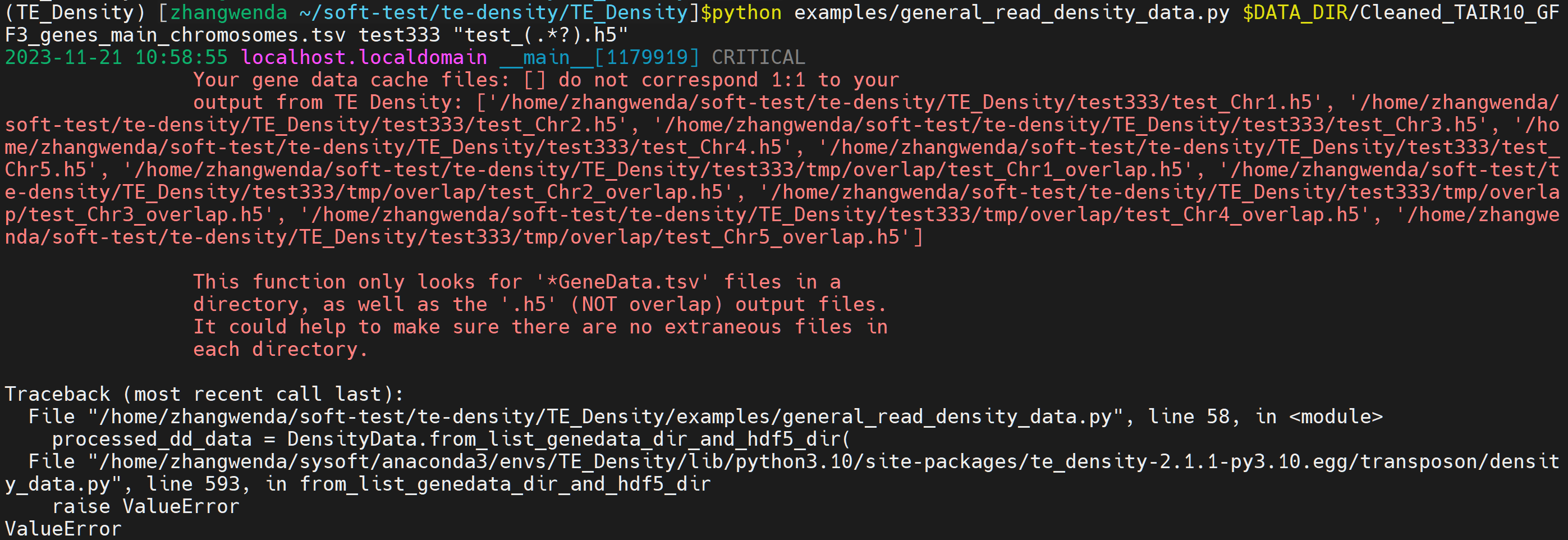
I tried to solve it, but this problem bothered me for many days. Can you give me some advice? What should I do and get the TEs density around the gene?
Thanks! Best wish to you!