 smorabit
commented
3 months ago
smorabit
commented
3 months ago Hi Dmitry,
I think that this is an interesting idea, but at this time I am not currently considering adding this as functionality to hdWGCNA. If you think that it makes sense for your data analysis, by all means proceed with this multi-module assignment of each gene.
I understand that with hdWGCNA or WGCNA you sometimes see genes in one module that have a very high kME in another module. Remember that genes are assigned to each module based on the a hierarchical clustering approach (Dynamic Tree Cut algorithm) applied to the co-expression topological overlap matrix (TOM). Based on this approach, genes are assigned to a single co-expression module. After this, we compute the eigengene-based connectivities (kMEs) by correlating the module eigengenes (MEs) with the expression of each gene. We can consider kMEs as a sort of "soft assignment" to complement the more strict assignment. There are alternative approaches for identifying gene modules, like topic modeling, which only use this kind of soft assignment approach, where each gene is weighted based on its contribution to each gene expression topic.
 Dimmiso
Dimmiso
Hi, thanks a lot for developing and supporting hdWGCNA!
As I understand functioning of hdWGCNA, assignment of genes to modules are strictly non-redundant: i.e. one gene can belong to only one module. However one gene may have quite high correlation coefficient (R) to two and more modules. I bumped on situation when a gene has R2 like 0.59, 0.58 and 0.55 to three different modules. If we think about modules and transcription regulones, it would make good sense to assign the same gene to two or more modules.
Such non-redundent gene assignment has one additional advantage, I think. It allows using such metrics as "ratio of uniquely assigned genes in a module" which strongly indicates module robustness. I tried such home-made procedure of non-redundent genes assignment. Plot below shows for each module proportion of genes which are 1) uniquely assigned (red), 2) green: non-uniquely but primary assigned (i.e. a gene has highest R for given module but it is also assigned to some other module(s) 3) blue: secondary assignment: a gene is assign to given module, but correspondent R is not the highest for given gene.
Do you have any comments on such non-redundent assignment? Is there such possibility in hdWGCNA or do you have plans to implement this?
Thanks a lot for supporting & developing hdWGCNA, a very helpful tool!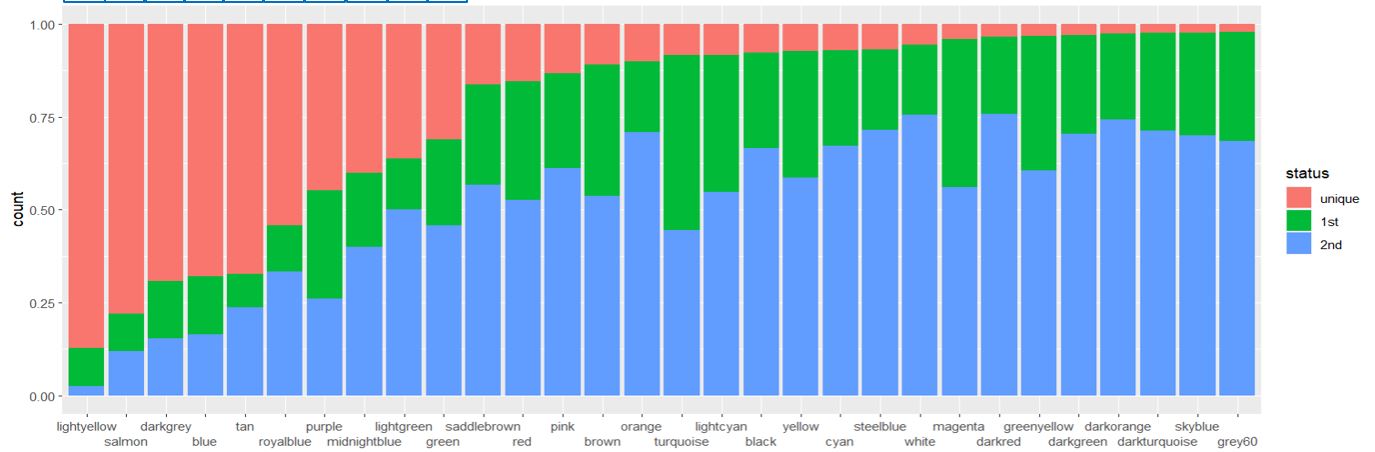
Dmitry