 martin-steinegger
commented
1 year ago
martin-steinegger
commented
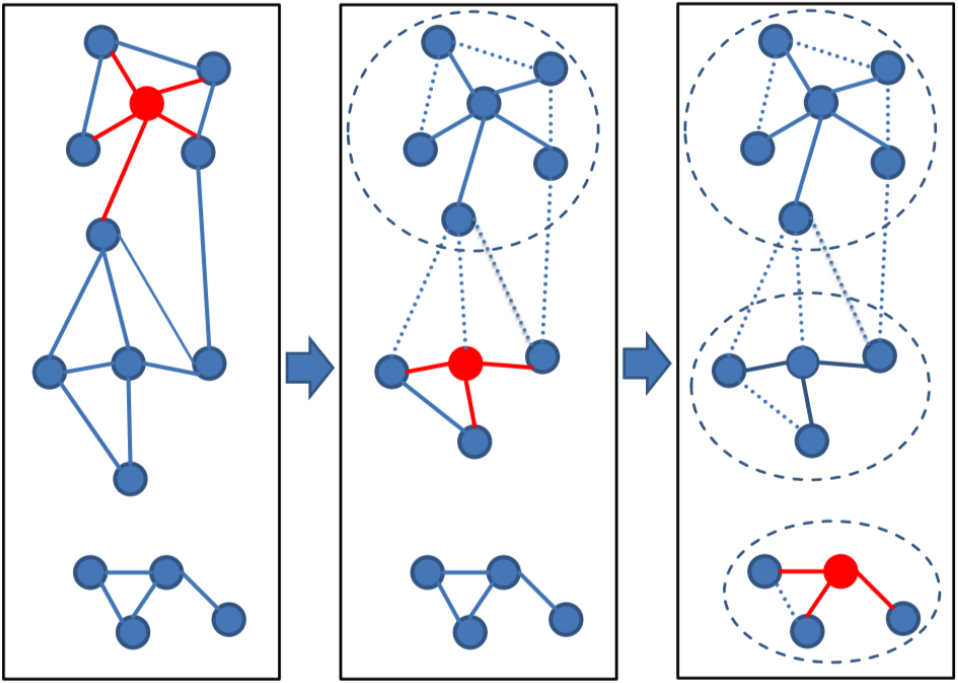
1 year ago @taylorreiter In default we use the greedy set cover algorithm for clustering. It picks the representative with the most alignments as the center. See the figure below. We use the longest sequence if you switch the --cov-mode 1 to uni-directional.
 milot-mirdita
milot-mirdita taylorreiter
taylorreiter
If I use the command:
How will mmseqs determine the representative cluster? I've tried reading through the documentation, issues, and papers, but it wasn't clear to me how the representative was selected for
easy-cluster...I'm sorry if I missed something! I think in the linclust paper it's mentioned that the longest sequence is selected as the representative, but I couldn't find a similar citation foreasy-cluster.