 seanb80
commented
7 years ago
seanb80
commented
7 years ago First sample finished in BSMap, 1 hour 26 minutes to run, 12.3% mapping efficiency.
Closed sr320 closed 7 years ago
seanb80
commented
7 years ago First sample finished in BSMap, 1 hour 26 minutes to run, 12.3% mapping efficiency.
seanb80
commented
7 years ago BSMap finished, starting Methratio now.
 sr320
commented
7 years ago
sr320
commented
7 years ago Here is some data from samples via CoGe https://genomevolution.org/coge/NotebookView.pl?nid=1888
seanb80
commented
7 years ago Methylation and Coverage histograms, Dendrogram, and PCA for just CpG sites.
Want me to start trimming things and re-running it?
 hputnam
commented
7 years ago
hputnam
commented
7 years ago Yes, please! I am just at this point with the CoGe results of bismark, so will compare.
sr320
commented
7 years ago I think there might be special trim consideration for RRBS? - I think Trim Galore might have special flag?
For the record this is RRBS. with MSPI as cut? is this a strand specific library?
hputnam
commented
7 years ago @seanb80 do you have your filtering cutoffs and treatment info listed somewhere? There are 3 treatments in this group.
hputnam
commented
7 years ago Yes, RRBS library cut with MSPI. Not strand specific.
seanb80
commented
7 years ago Filtering Cutoffs are Min Coverage of 3, high Percentage of 95. As far as treatment info, I've got it coded as Ambient = 0, Non-Ambient = 1, according to the Methylkit documentation, the treatment argument only accepts a vector containing 0 and 1.
Sorry I don't have notebooks for this stuff, Realized tonight I neglected to install R Studio Server, so I'm just programming in the console at the moment.
I'm using the -rrbs option in trim galore btw.
Call for a single trim galore run looks like
/home/shared/trimgalore/trim_galore --fastqc -q 20 --rrbs --paired --length 20 EPI-145_S38_L005_R1_001.fastq.gz EPI-145_S38_L005_R2_001.fastq.gz -o ~/Documents/Geoduck/trimmed-data
Does that look right?
hputnam
commented
7 years ago It will accept 1,2,3 which will allow it to color 3 different treatments, but not sure what is happening behind the scenes... trying to post my notebook now
seanb80
commented
7 years ago Ok, I can change that real quick and re-upload stuff.
seanb80
commented
7 years ago hputnam
commented
7 years ago Thanks! ... I have frozen my repo with large files and will post notebook for comparison as soon as I troubleshoot. Happy new year from the east coast!
hputnam
commented
7 years ago seanb80
commented
7 years ago Trimming and FastQC done, data and qc reports available at owl.fish.washington/web/scaphapoda/Sean/geoduck-trimmed-data.
BSMap started, will update when that's done (probably tomorrow?)
seanb80
commented
7 years ago Hmm, or not. Getting a buffer overflow error from BSMap. Not sure what's causing it but will do some research.
hputnam
commented
7 years ago Do you have any bedgraph or gff files from the initial run?
seanb80
commented
7 years ago I don't at the moment, but I can figure out how to make them from bsmap results when I get home in a few hours.
On Jan 1, 2017, at 12:29 PM, Hollie Putnam notifications@github.com wrote:
Do you have any bedgraph or gff files from the initial run?
— You are receiving this because you were mentioned. Reply to this email directly, view it on GitHub, or mute the thread.
sr320
commented
7 years ago I am generating bams now that can be visualized in IGV http://d.pr/i/RKal
Bams are @ http://owl.fish.washington.edu/halfshell as they become available. as is genome...
Then making begraphs of bams.
Are methratio files are available on owl? - I can convert easily to begraphs
!grep "[A-Z][A-Z]CG[A-Z]" </Volumes/caviar/wd/2016-11-14c/methratio_out_{i}.txt> \
/Volumes/caviar/wd/2016-11-14c/methratio_out_{i}_CG.txt
%%bash
awk '{if ($8 >= 3) print $1,$2-1,$2+1,"CpG",$5}' \
/Volumes/caviar/wd/2016-11-14c/methratio_out_1_ATCACG_CG.txt | tr ' ' "\t" \
> /Volumes/caviar/wd/2016-11-14c/1_ATCACG.igv
Here is a session file to see some of the BAMs in IGV
http://owl.fish.washington.edu/halfshell/igv_session-EPI-010117.xml
seanb80
commented
7 years ago Copying the files to here now. the CPG_ files are just the CpG motifs, the methratio-out- files are raw methratio files.
http://owl.fish.washington.edu/scaphapoda/Sean/geoduck-untrimmed-data/
I'm having a problem with the trimmed files with BSmap, I'm getting a segmentation fault when it tries to load the second file. I've tried unzipping them to the raw FQ files to no avail, but will continue working on it.
sr320
commented
7 years ago IGV files are writing out now to owl
just reporting loci with at least 10x coverage
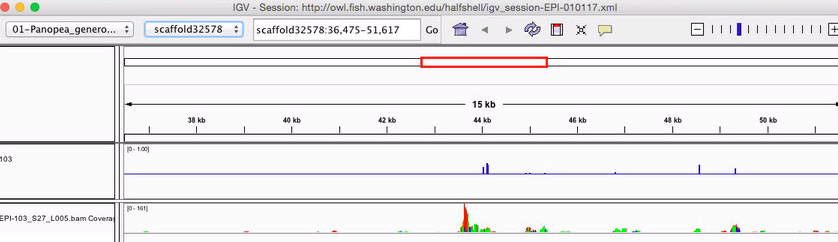
http://owl.fish.washington.edu/halfshell/index.php?dir=&sort=date&order=desc
hputnam
commented
7 years ago Seems to reflect same patterning as clustering/PCA... no strong treatment differences visible, which would also match the physiology.

sr320
commented
7 years ago Here are some random pics of loci that might be considered different between treatment.. Might be worth putting up pic above (all on Igv) and some select differences on poster to show no dramatic changes, but we are looking at individual loci such as these....
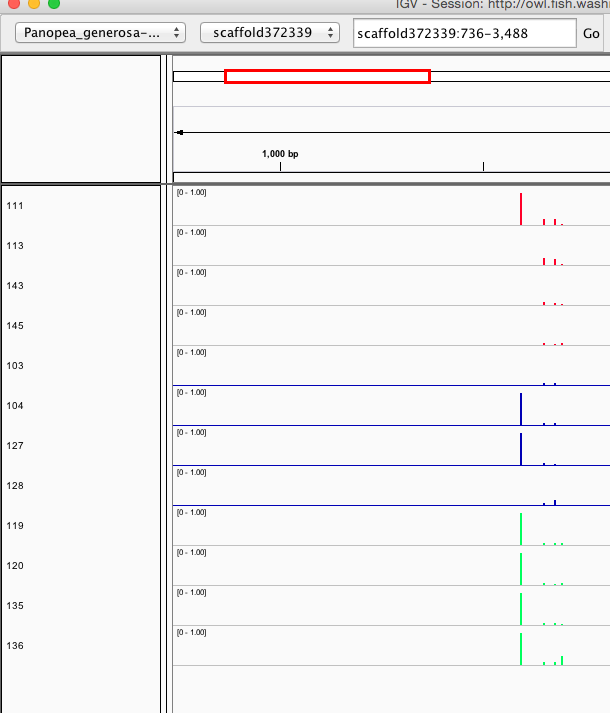
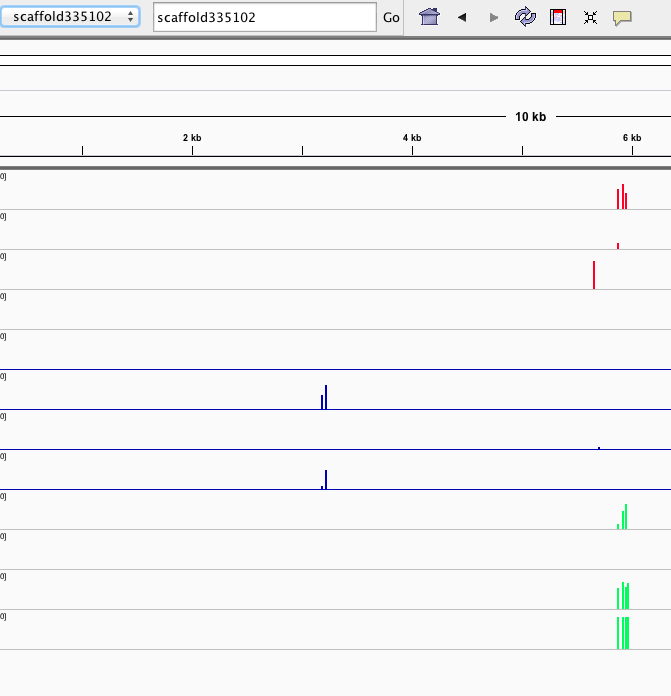

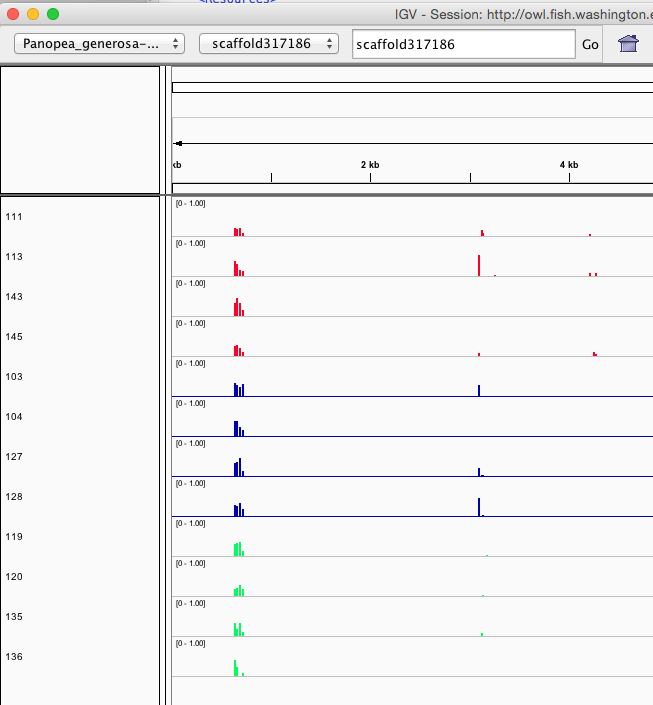
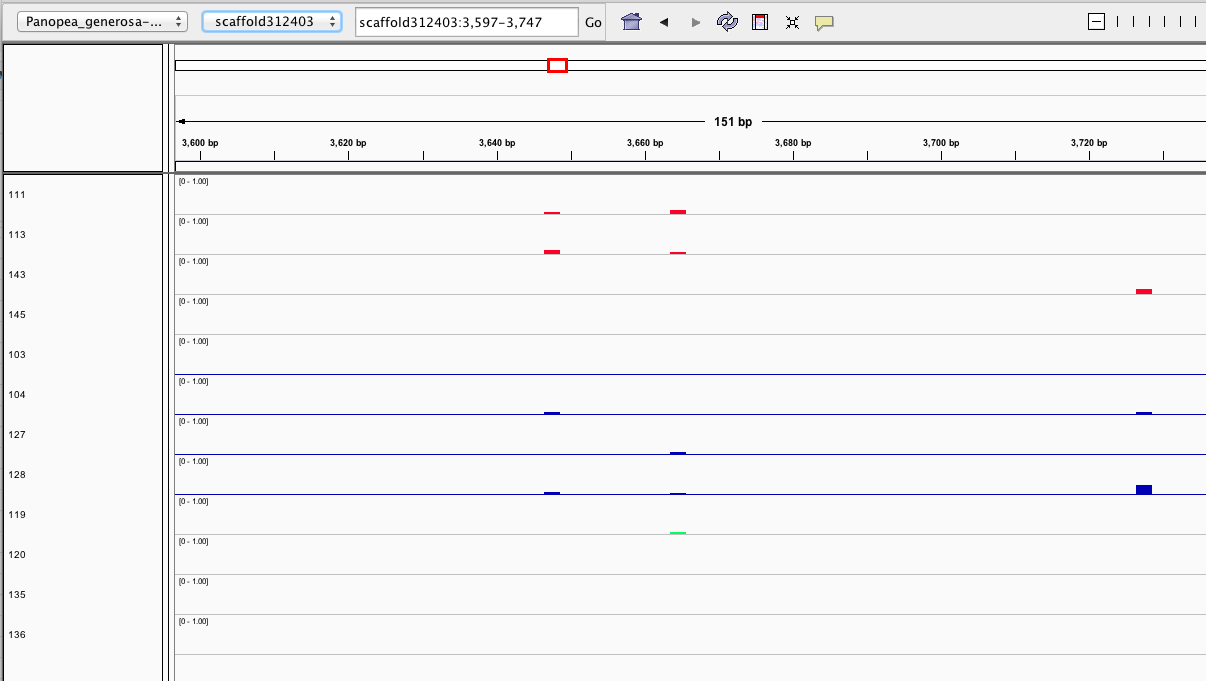

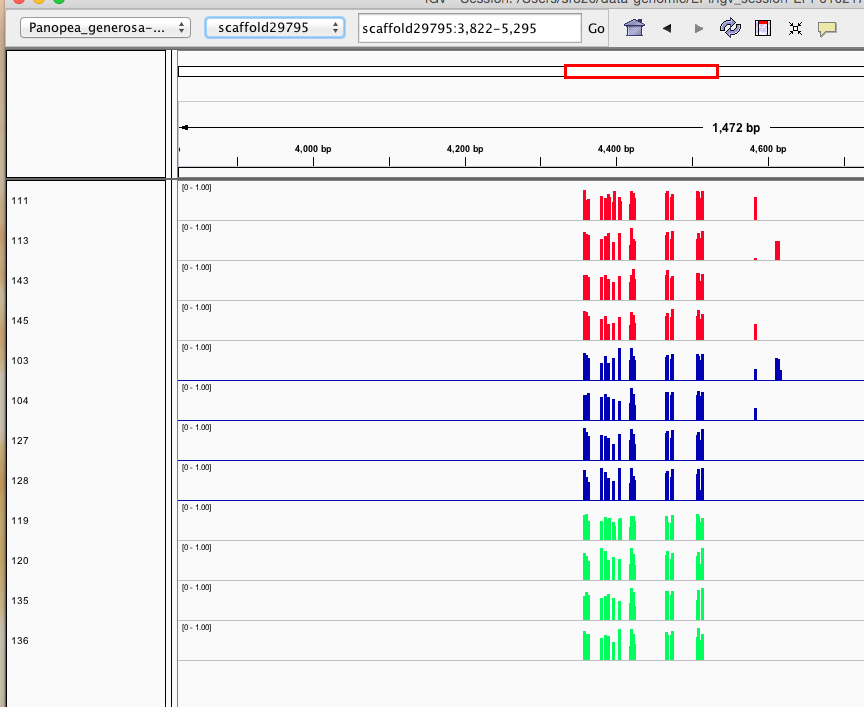
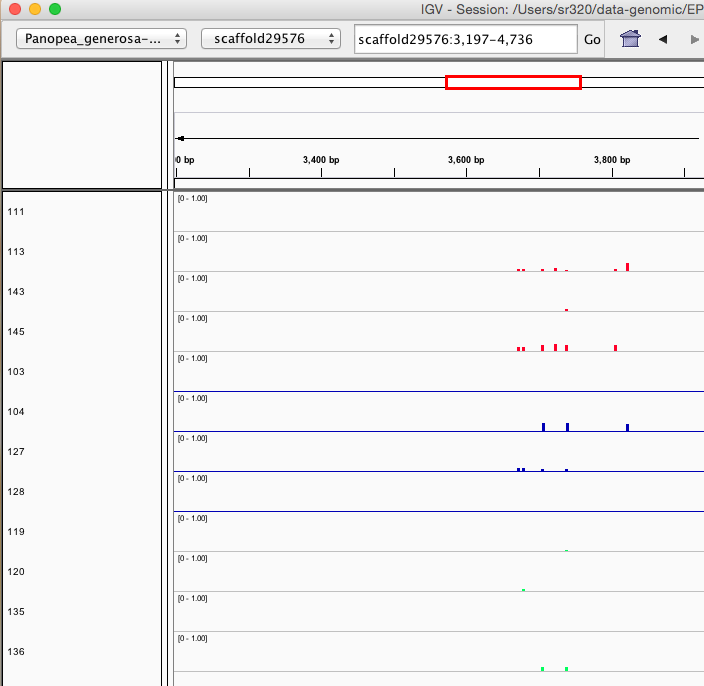
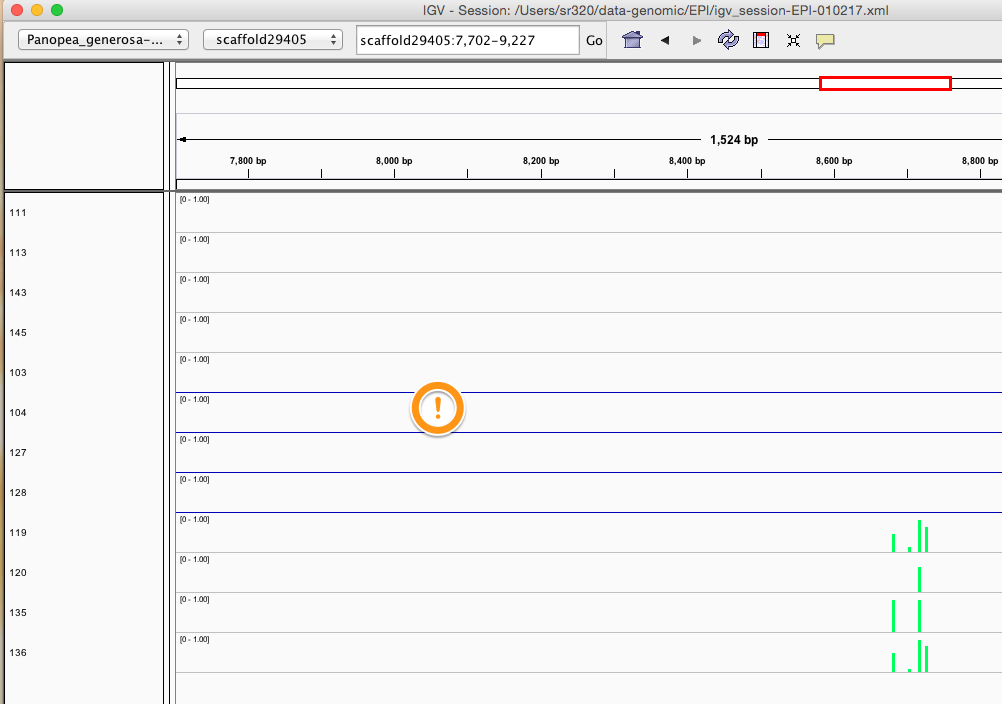

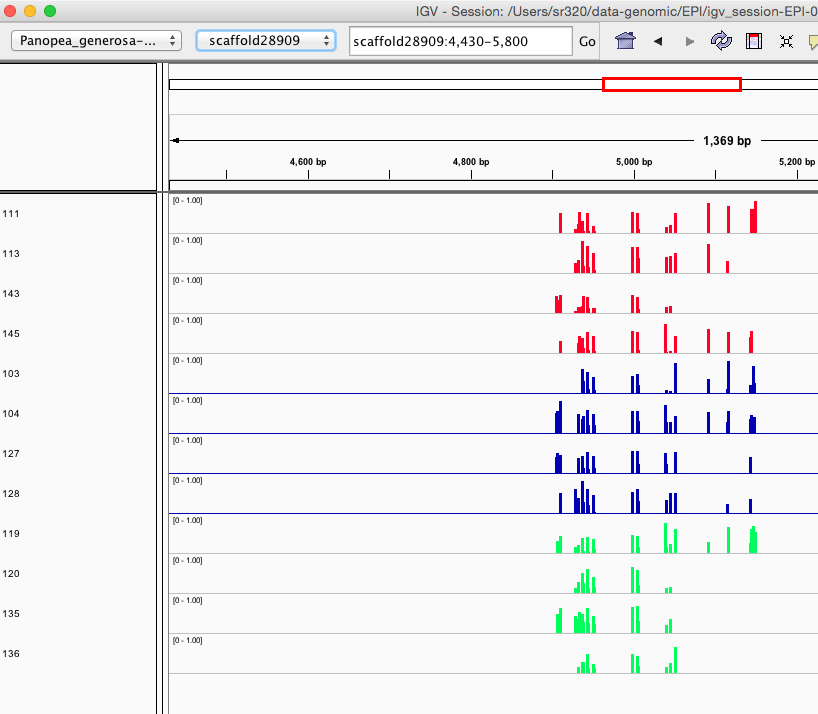
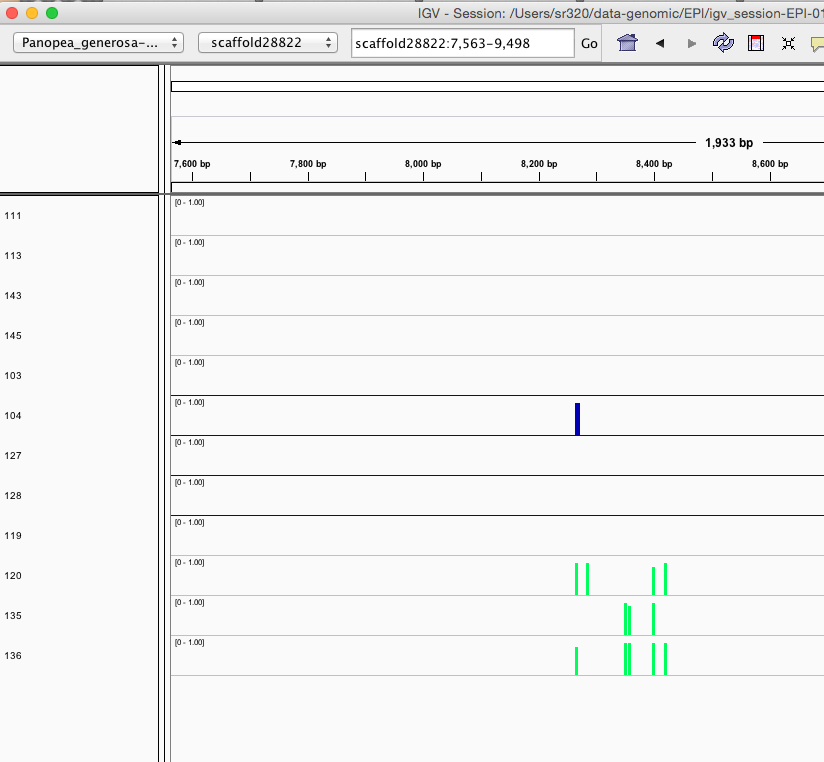
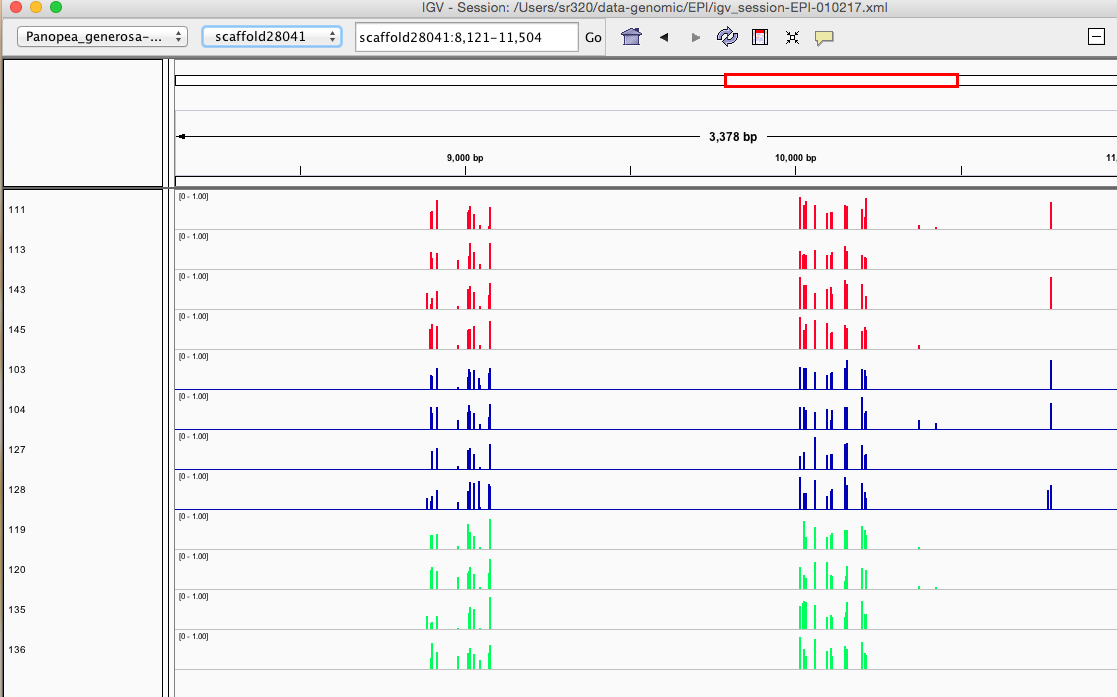
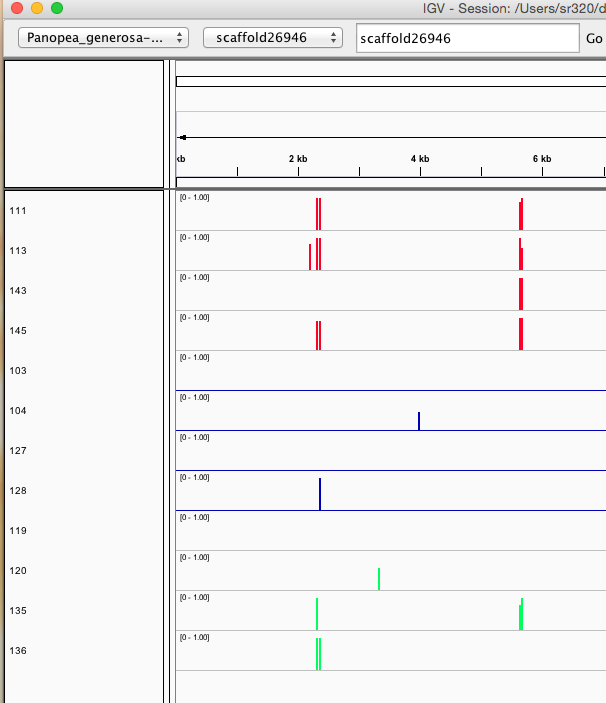
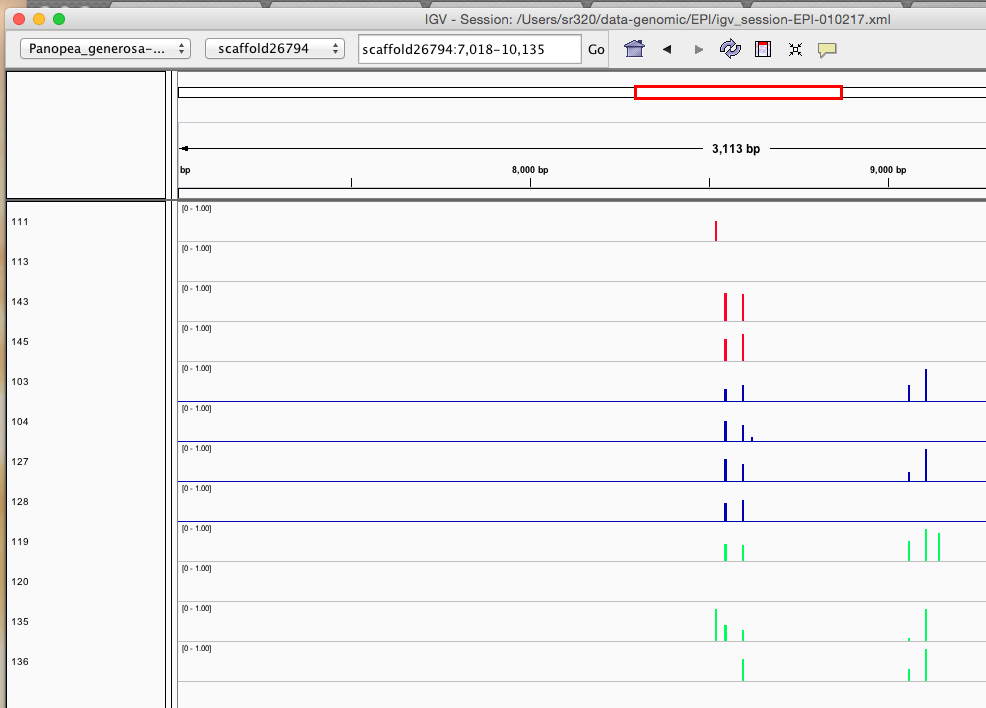
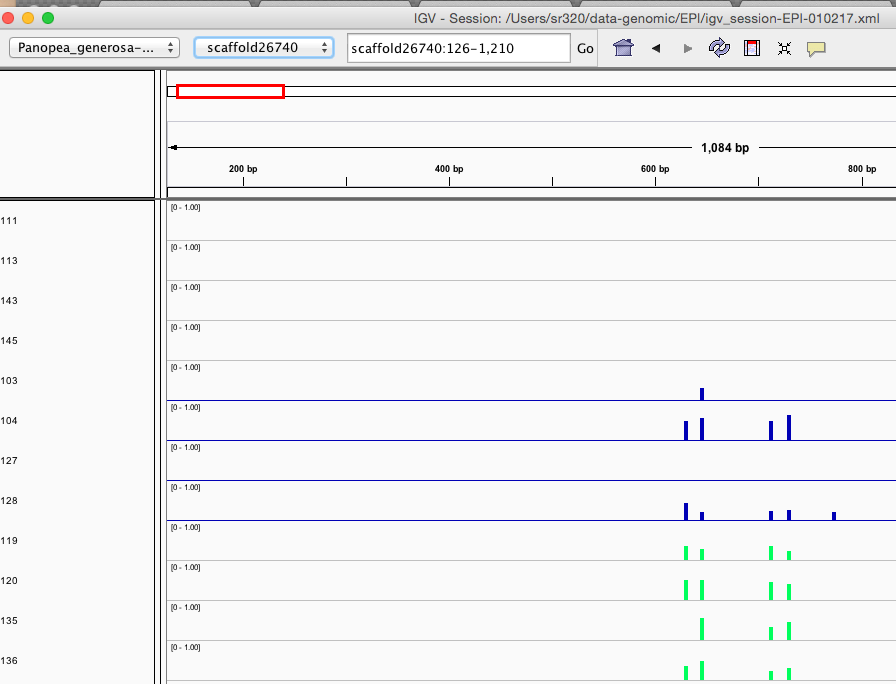
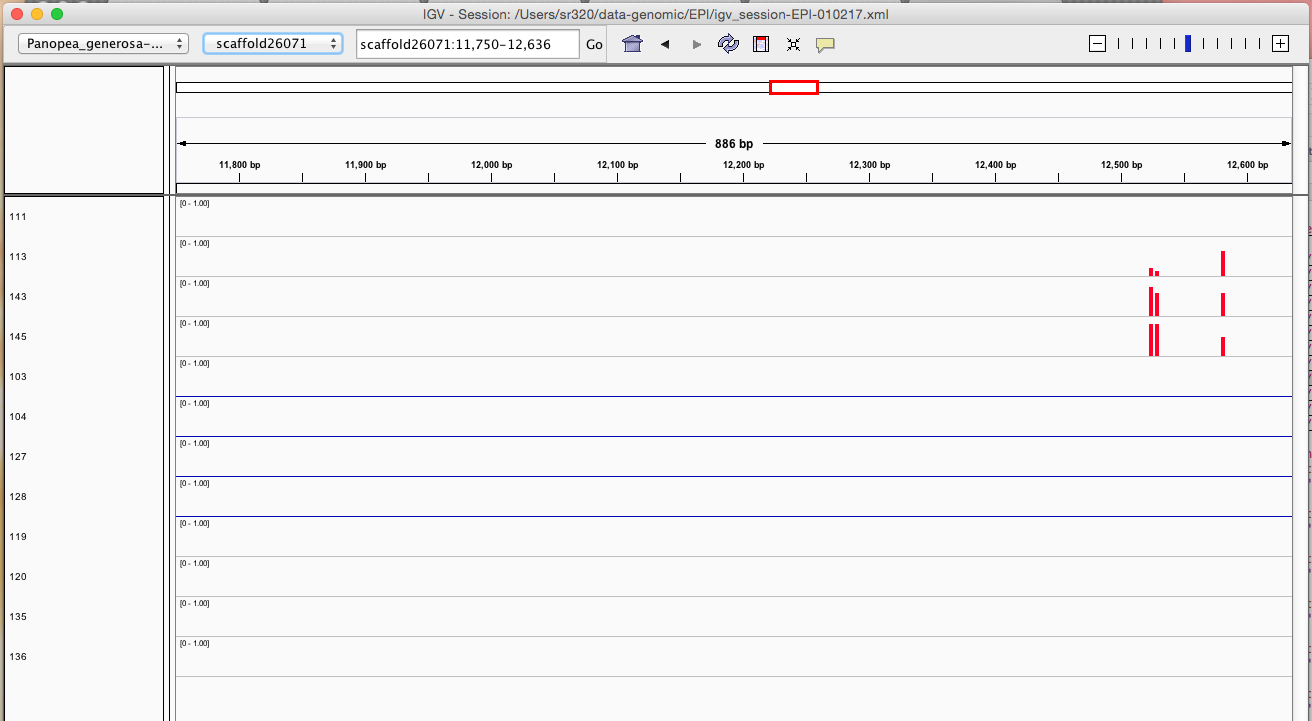 hputnam
commented
7 years ago
hputnam
commented
7 years ago Is there a place on campus to get the poster printed tomorrow? Or should I plan for a Kinkos?
sr320
commented
7 years ago Yes - I think this place https://f2.washington.edu/fm/c2/posters
though I have never done one. @kubu4 is this what we normally do?
 kubu4
commented
7 years ago
kubu4
commented
7 years ago Yes, that's it. The last time we had them print a poster, they were able to do it same-day. I'd recommend getting the poster submitted sometime today. This way, they'll likely have a proof ready for review in the morning that I can walk over to view/approve, and then have the print finished by 5PM.
seanb80
commented
7 years ago Finally got trimmed stuff through BSmap and Methylkit, for some reason the trimmed .gz files caused a buffer overrun in BSmap, but the uncompressed .fq files didn't. Doesn't look much different than the bismark or untrimmed data.
output here
Will upload raw data files to Owl here shortly.
sr320
commented
7 years ago Thanks!
Maybe start with bsmap to get through to data/figures, then go back and hit other pipelines.
Timeline wise - looking to have something preliminary next week.