 sr320
commented
6 years ago
sr320
commented
6 years ago Big picture, would it be correct to assume you are trying to get GO Slim terms for each protein? On Mon, Jul 10, 2017 at 8:55 PM Rhonda Elliott notifications@github.com wrote:
I've got a file where I am trying to unfold a column with multiple GO terms delimited by semicolons. I have performed this function many times in Galaxy but for whatever reason I cannot get it to work for me right now.
Here is the file https://github.com/RobertsLab/project-pacific.oyster-larvae/blob/master/DIA_2015/AnnotatedproteinsGO.tabular
It's driving me bonkers. Does anyway have an easy work around or see something that might be my problem? I just tried redoing a file I had successfully done in the past and I got the same error.
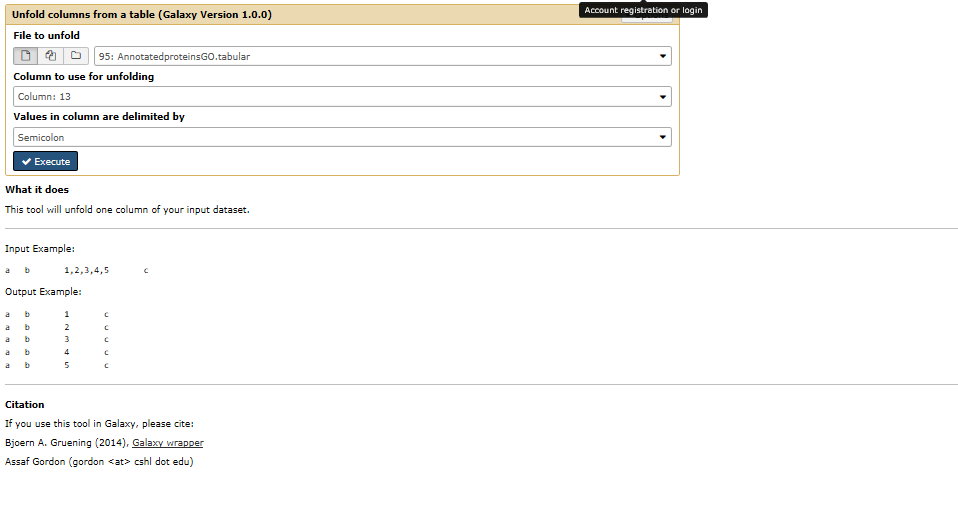
This is what I am trying: [image: image] https://user-images.githubusercontent.com/20071030/28049813-9ea10586-65ae-11e7-8884-69e8455f0371.png
And this is what I get: [image: image] https://user-images.githubusercontent.com/20071030/28049841-cc04d548-65ae-11e7-8bb3-0b4955b87d64.png
I tried emailing them but have no idea what their turn around is for responding, a day, a week, a month?
— You are receiving this because you are subscribed to this thread. Reply to this email directly, view it on GitHub https://github.com/sr320/LabDocs/issues/654, or mute the thread https://github.com/notifications/unsubscribe-auth/AEPHt4HDMLgRUsaMNylQRNn52POfD_Sdks5sMvH8gaJpZM4OTsjd .
 Ellior2
Ellior2
 kubu4
kubu4



{kind=link}
{kind=link}
I've got a file where I am trying to unfold a column with multiple GO terms delimited by semicolons. I have performed this function many times in Galaxy but for whatever reason I cannot get it to work for me right now.
Here is the file
It's driving me bonkers. Does anyway have an easy work around or see something that might be my problem? I just tried redoing a file I had successfully done in the past and I got the same error.
This is what I am trying:
And this is what I get:
I tried emailing them but have no idea what their turn around is for responding, a day, a week, a month?