 kubu4
commented
2 years ago
kubu4
commented
2 years ago Just a heads up, since these are already bisulfite converted reads, the taxonomic assignments are probably going to be somewhat inaccurate.
Closed sr320 closed 4 months ago
kubu4
commented
2 years ago Just a heads up, since these are already bisulfite converted reads, the taxonomic assignments are probably going to be somewhat inaccurate.
kubu4
commented
2 years ago I was starting to work on this and noticed that some of the files had time stamps listed as this morning. Are these still part of an active Mox job (i.e. should I wait to start this)?
 sr320
commented
2 years ago
sr320
commented
2 years ago It can wait
sr320
commented
2 years ago It can wait
kubu4
commented
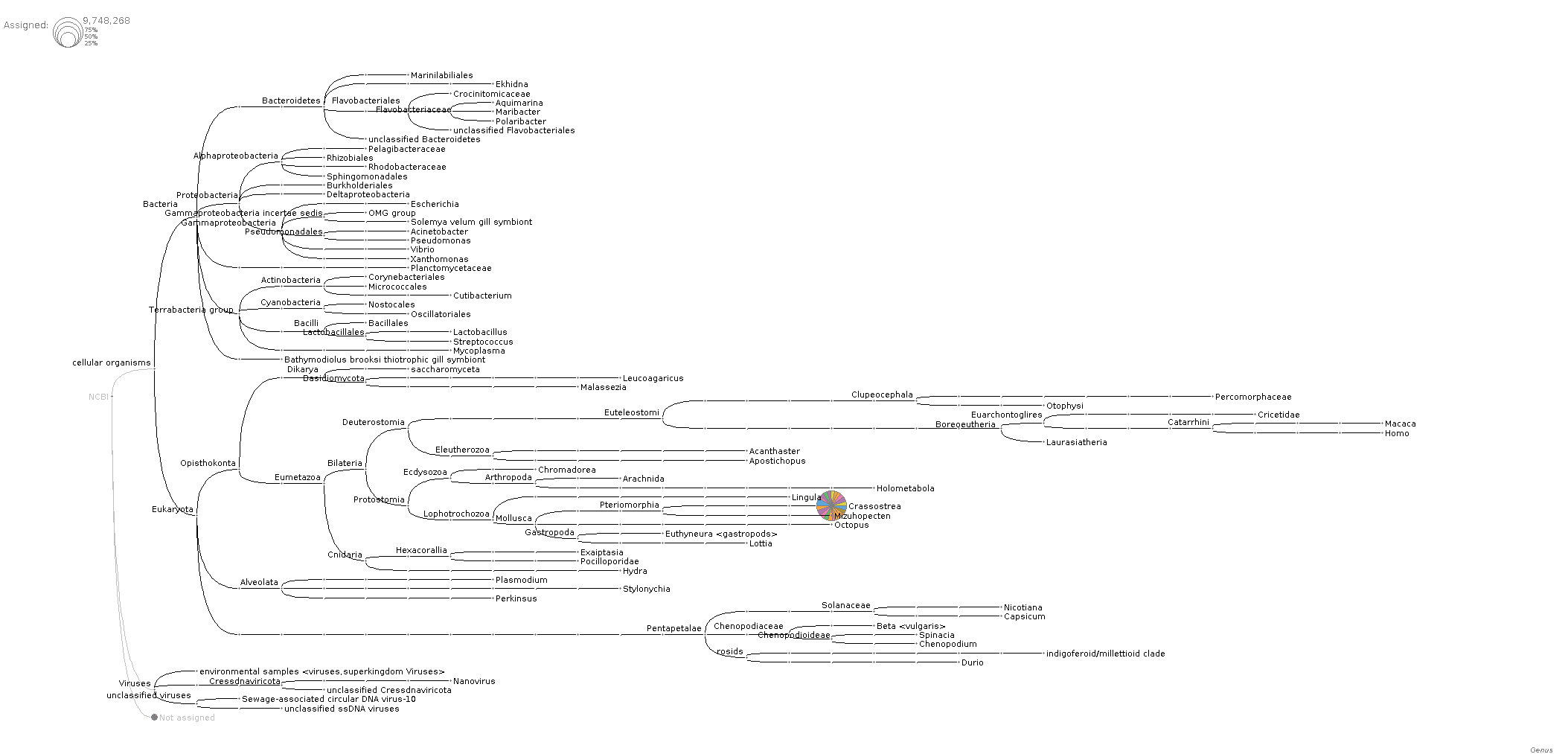
2 years ago Looks like bulk of reads (i.e. > 75%) map to Crassostrea (see images at bottom of post).
So, not sure why they're not getting mapped...
I'm assuming these came from Bismark? If yes, the manual provides a bit of insight into capturing unmapped reads. Additionally, a comparison of unmapped reads and ambiguous reads might allow a better grasp of what's happening:
--unWrite all reads that could not be aligned to the file _unmapped_reads.fq.gz in the output directory. Written reads will appear as they did in the input, without any translation of quality values that may have taken place within Bowtie or Bismark. Paired-end reads will be written to two parallel files with _1 and _2 inserted in their filenames, i.e.
unmapped_reads_1.fq.gzandunmapped_reads_2.fq.gz. Reads with more than one valid alignment with the same number of lowest mismatches (ambiguous mapping) are also written to unmapped_reads.fq.gz unless--ambiguousis also specified.
--ambiguousWrite all reads which produce more than one valid alignment with the same number of lowest mismatches or other reads that fail to align uniquely to _ambiguous_reads.fq. Written reads will appear as they did in the input, without any of the translation of quality values that may have taken place within Bowtie or Bismark. Paired-end reads will be written to two parallel files with _1 and _2 inserted in their filenames, i.e.
_ambiguous_reads_1.fqand_ambiguous_reads_2.fq. These reads are not written to the file specified with--un.
Wordcloud of taxonomic assignments of reads for each sample (Genus level):

Phylogenetic tree of taxonomic assignments of reads for each sample (Genus level):
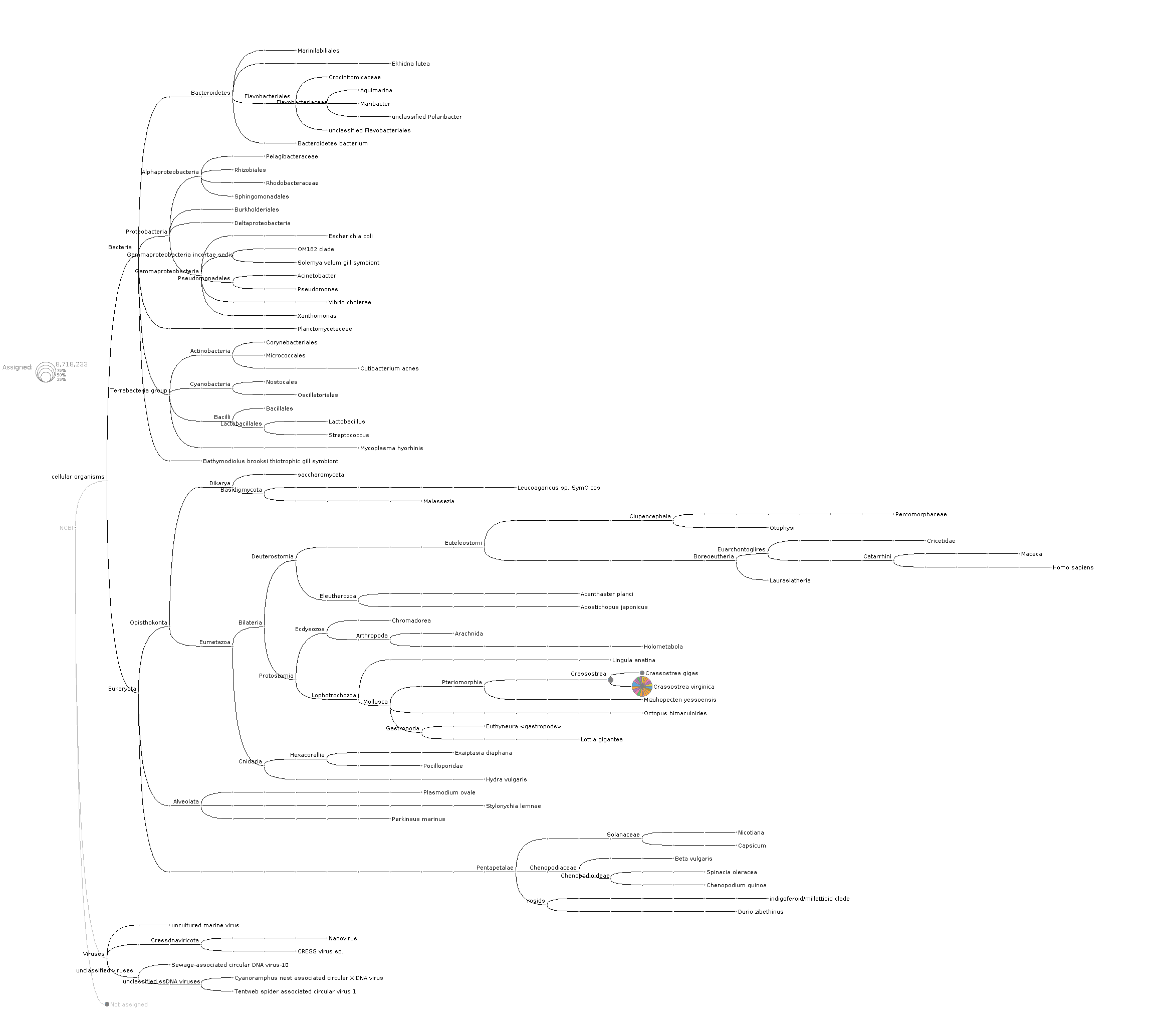
And, here's the tree at the species level to help confirm that there's not a bunch of C.gigas contamination:
Discussed in https://github.com/sr320/ceabigr/discussions/11
@kubu4 can you given MEGAN a whirl on unmapped BS reads?
Currently they are located at
/gscratch/scrubbed/sr320/021022-BS-unmap/*unmapped_reads*.fq.gz