 pangxueyu233
commented
2 years ago
pangxueyu233
commented
2 years ago Hi Tan,
I found the chromosome name of my data had 'chr', so I modified the source data of reg3.py to make it possible to remove the CN regions as following:
......
# presets
presets = {
"hm":[Reg("chr" + str(i + 1)) for i in range(22)] + [Reg("X"), Reg("Y")],
"hf":[Reg("chr" + str(i + 1)) for i in range(22)] + [Reg("X")],
"mm":[Reg("chr" + str(i + 1)) for i in range(19)] + [Reg("chrX"), Reg("chrY")],
"mf":[Reg("chr" + str(i + 1)) for i in range(19)] + [Reg("chrX")]}
preset_descriptions = {
"hm":"human male",
"hf":"human female",
"mm":"mouse male",
"mf":"mouse female"}
......But when I used the dip-c reg3 to exclude the CN regions in .3dg files, it would return errors. The codes I used are as following:
DIP_C=./programme/dip-c-master/dip-c
sample=MLL3_RS2_1
rep=1
$DIP_C reg3 -e $sample.filter.haplotype.bed -p hf $sample.20k.${rep}.3dg > $sample.20k.${rep}.filter.3dgAnd it returns:
Traceback (most recent call last):
File "/mnt/data/user_data/xiangyu/programme/dip-c-master/dip-c", line 130, in <module>
main()
File "/mnt/data/user_data/xiangyu/programme/dip-c-master/dip-c", line 69, in main
return_value = reg3.reg3(sys.argv[1:])
File "/mnt/data/user_data/xiangyu/programme/dip-c-master/reg3.py", line 89, in reg3
g3d_data = file_to_g3d_data(open(args[0], "rb"))
File "/mnt/data/user_data/xiangyu/programme/dip-c-master/classes.py", line 1427, in file_to_g3d_data
g3d_data.add_g3d_particle(string_to_g3d_particle(g3d_file_line.strip()))
File "/mnt/data/user_data/xiangyu/programme/dip-c-master/classes.py", line 1214, in string_to_g3d_particle
hom_name, ref_locus, x, y, z = g3d_particle_string.split("\t")
ValueError: need more than 1 value to unpackAnd I also checked the source data of reg3.py, the error happed in this function file_to_g3d_data. And when I execute the following codes, it would reported same errors:
import sys
import getopt
import gzip
from classes import Reg, file_to_reg_list, file_to_g3d_data, G3dData
g3d_data = file_to_g3d_data(open("./MLL3_RS2_1.20k.1.3dg", "rb"))it returns:
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
File "classes.py", line 1427, in file_to_g3d_data
g3d_data.add_g3d_particle(string_to_g3d_particle(g3d_file_line.strip()))
File "classes.py", line 1214, in string_to_g3d_particle
hom_name, ref_locus, x, y, z = g3d_particle_string.split("\t")
ValueError: need more than 1 value to unpackCan you help me work it out?
And I had submitted my .3dg file and abnomal CN region data.
mydata.zip
Thanks Xiangyu Pan
 tanlongzhi
tanlongzhi

Hi Tan,
it's really a interesting tool to analyse the 3D genome on single-cell level. Recently, we also implemented this platform in our projects. But we met some problems. As you can see in some cells of our data, there were obvious copy-number (CN) gains or losses regions. Although you mentioned
.regfiles inREADME.mdfile, I'm not sure where and how should we do to remove them.Now, I have two plans to achive it: 1) remove the CN regions by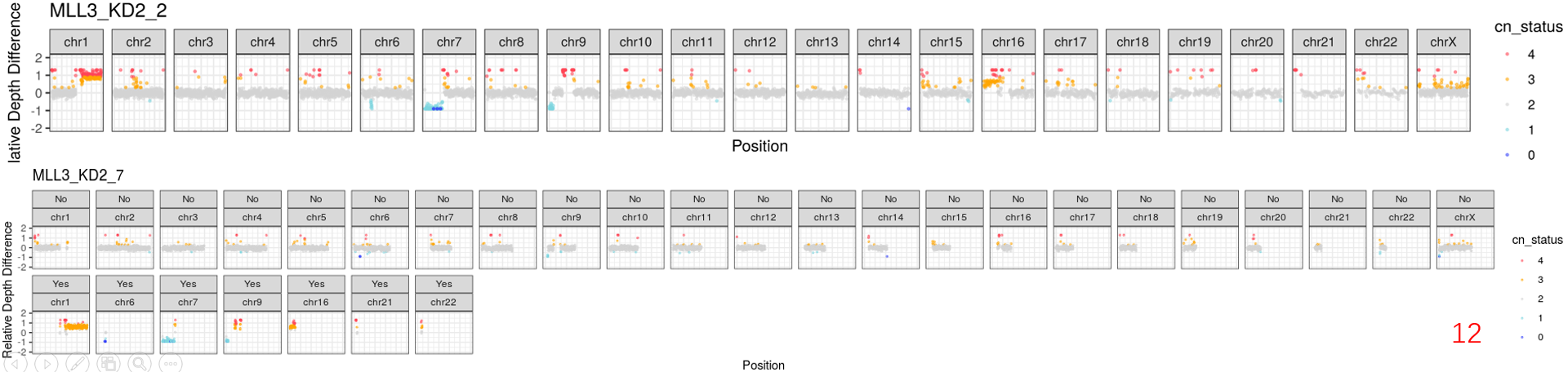
samtoolsandbedtoolsfrom*.aln.sam.gzfiles; 2) remove them bydip-c reg3. But I'm not sure which way is better, because theRMSDis the important standard for the structure. And theRMSDscores would be calculated based on*3dgfiles. So, I prefer to remove these region by the first way to before genome structure reconstruction. Do you think whether it's resonbale to do?Sincerely Xiangyu Pan West China hospital Sichuan University Chengdu, China