DBSCAN-SWA: an integrated tool for rapid prophage detection and annotation
Table of Contents
- Background
- Requirements
- Install
- Usage
- Visualizations
- Contributors
- License
Background
Bacteriophages are viruses that specifically infect bacteria and the infected bacteria are called bacterial hosts of the viruses. Passive replication of the bacteriophage genome relies on integrate into the host's chromosome and becoming a prophage. Prophages coexist and co-evolve with bacteria in the natural environment, having an impact on the entire ecological environment. Therefore, it is very essential to develop effective and accurate tools for identification of prophages. DBSCAN-SWA, a command line software tool developed to predict prophage regions in bacterial genomes, running faster than any previous tools and presenting great detection power based on the analysis using 184 manually curated prophages
Requirements
The source code is written by python3. In addition, several tools have been applied in DBSCAN-SWA. Among these, Prokka requires installtion by users.
First, please install the following python packages:
-
numpy
-
Biopython
-
sklearn
Second, please install the following tools:
- Prokka in https://github.com/tseemann/prokka
git clone https://github.com/tseemann/prokka.git # install the dependencies: sudo apt-get -y install bioperl libdatetime-perl libxml-simple-perl libdigest-md5-perl # install perl package sudo bash export PERL_MM_USE_DEFAULT=1 export PERL_EXTUTILS_AUTOINSTALL="--defaultdeps" perl -MCPAN -e 'install "XML::Simple"' # install the prokka databases prokka --setupdb # test the installed prokka databases prokka --listdbwarning: Prokka needs blast+ 2.8 or higher, so we provide the version of blast+ in bin directory, the users can install a latest blast+ and add it to the environment or use the blast+ provided by DBSCAN-SWA. Please ensure the usage of blast+ in your environment by eg:
which makeblastdb
Install
Linux
- step1: Download the whole packages and partial profiles from https://github.com/HIT-ImmunologyLab/DBSCAN-SWA
git clone https://github.com/HIT-ImmunologyLab/DBSCAN-SWA - step2: Download DBSCAN-SWA database for standalone from webserver
When the DBSCAN-SWA program is run for the first time, it will download the required databases by default, or you can download the databases manually by setting --download_db' to manual. There are two ways to download the database manually, the first one is from DBSCAN-SWA server and the second one is from Zenodo.
### Download database from DBSCAN-SWA server
wget -c -b http://www.microbiome-bigdata.com/PHISDetector/static/download/DBSCAN-SWA/db.tar.gz
### Access dabase from Zenodo
https://zenodo.org/records/10404224- step3: Unzip the database file to specified subdirectory under DBSCAN-SWA installation directory
### Unzip the database file
tar -zxvf path/to/db.tar.gz
### Put the unzipped database files in specified subdirectory
cp path/to/download/db path/to/DBSCAN-SWA- step4: Add the [download_path]/bin to your environment.
export PATH=$PATH:/path/to/DBSCAN-SWA/software/blast+/bin:$PATH
export PATH=$PATH:/path/to/DBSCAN-SWA/bin
export PATH=$PATH:/path/to/DBSCAN-SWA/software/diamond
export PATH=$PATH:/path/to/prokka/bin- step5: Grant permission to run the softwares.
chmod u+x -R /path/to/DBSCAN-SWA/bin chmod u+x -R /path/to/DBSCAN-SWA/software - step6: Test DBSCAN-SWA in command line
python <path>/dbscan-swa.py --hUsage
DBSCAN-SWA is an integrated tool for detection of prophages, providing a series of analysis including ORF prediction and genome annotation, phage-like gene clusters detection, attachments site identification and infecting phages annotation
Command line options
General:
--input <file name> : Query phage file path: FASTA or GenBank file
--output <folder name> : Output folder in which results will be stored
--prefix <prefix> : default: bac:
Phage Clustering:
--evalue <x> : maximal E-value of searching for homology virus proteins from viral UniProt TrEML database. default:1e-7
--min_protein_num <x> : optional,the minimal number of proteins forming a phage cluster in DBSCAN, default:6
--protein_number <x> : optional,the number of expanding proteins when finding prophage att sites, default:10
Phage Annotation:
--add_annotation <options> : optional,default:PGPD,
1.PGPD: a phage genome and protein database,
2.phage_path:specified phage genome in FASTA or GenBank format to detect whether the phage infects the query bacteria
3.none:no phage annotation
--per <x> : Minimal % percentage of hit proteins on hit prophage region(default:30)
--idn <x> : Minimal % identity of hit region on hit prophage region by making blastn(default:70)
-cov <x> : Minimal % coverage of hit region on hit prophage region by making blastn(default:30)Start DBSCAN-SWA
The python script is also provided for expert users
1.predict prophages of query bacterium with default parameters:
python <path>/dbscan-swa.py --input <bac_path> --output <outdir> --prefix <prefix>- predict prophages of query bacterium and no phage annotation:
python <path>/dbscan-swa.py --input <bac_path> --output <outdir> --prefix <prefix> --add_annotation none - predict prophages of query bacterium and detect the bacterium-phage interaction between the query bacterium and query phage:
python <path>/dbscan-swa.py --input <bac_path> --output <outdir> --prefix <prefix> --add_annotation <phage_path>Outputs
| File Name | Description |
|---|---|
| \<prefix>_DBSCAN-SWA_prophage_summary.txt | the tab-delimited table contains predicted prophage informations including prophage location, specific phage-related key words, CDS number, infecting virus species by a majority vote and att sites |
| \<prefix>_DBSCAN-SWA_prophage.txt | this table not only contains the information in |
| all predicted prophage Nucleotide sequences in FASTA format | |
| all predicted prophage protein sequences in FASTA format | |
| Phage Annotation | if add_annotation!=none, the following files are in "prophage_annotation" |
| the tab-delimited table contains the information of prophage-phage pairs with prophage_homolog_percent, prophage_alignment_identity and prophage_alignment_coverage | |
| the table contains the detailed information of bacterium-phage interactions including blastp and blastn results |
Visualizations
You can find a directory named "test" in the DBSCAN-SWA package. The following visualzations come from the predicted results of Xylella fastidiosa Temecula1(NC_004556)
(1) the genome viewer to display all predicted prophages and att sites
 (2) the detailed information of predicted prophages
(2) the detailed information of predicted prophages
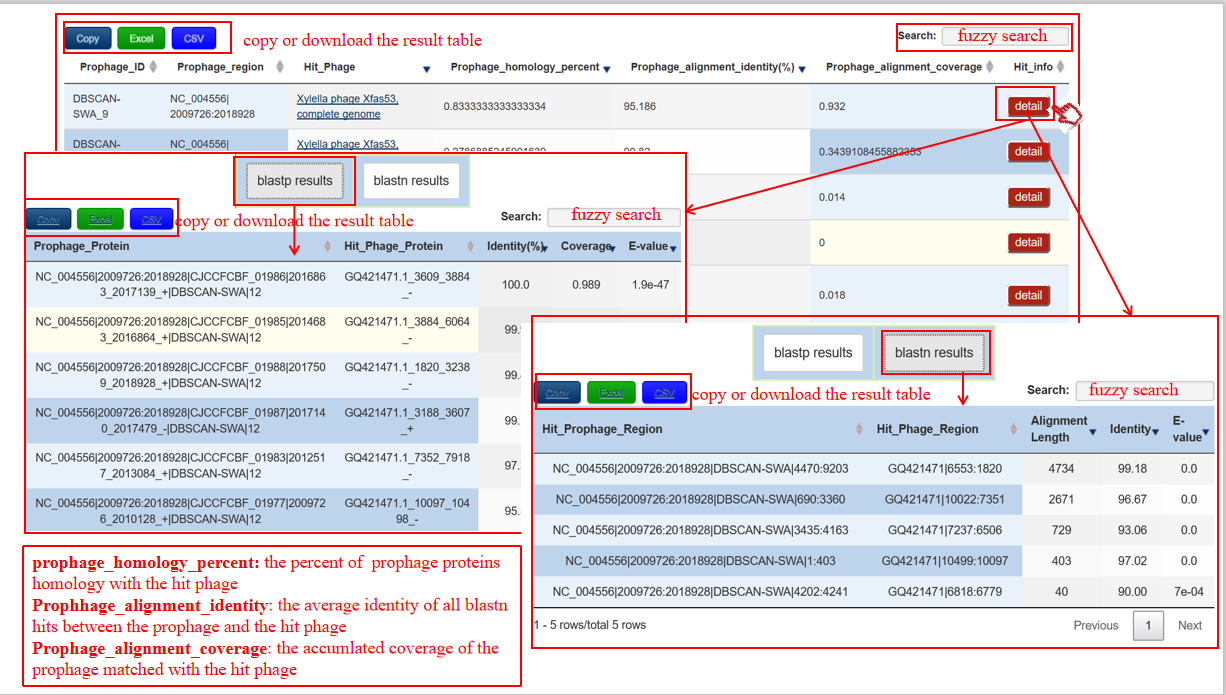
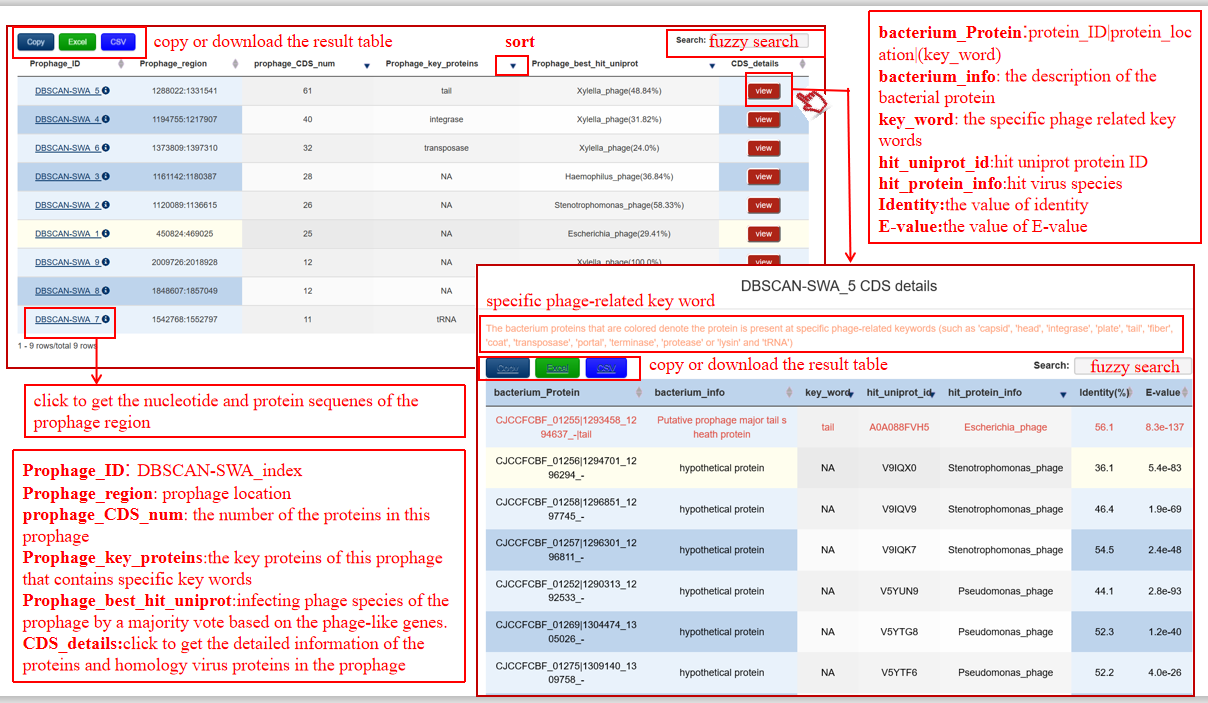 (3) If the users set add_annotation as PGPD or the phage file path, the detailed information of bacterium-phage interaction will be illustrated as follows:
(3) If the users set add_annotation as PGPD or the phage file path, the detailed information of bacterium-phage interaction will be illustrated as follows:
Contributors
This project exists thanks to all the people who contribute.
License
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.