BEPH
Official repo for XXXXX, which is based on BEiTv2:
*It is worth noting that the BEiT implementation we use comes from mmselfsup[https://github.com/open-mmlab/mmselfsup].
Key Features
This is the repo for the paper XXXXXXX led by ZhaochangYang and XXXXX:
- BEPH is pre-trained on 11 million histopathological images from TCGA with self-supervised learning
- BEPH has been validated in multiple cancer detection and survival prediction tasks
- BEPH can be efficiently adapted to customised tasks
Install environment
Install mmselfsup
conda create -n BEPH python=3.9 -y
conda activate BEPH
conda install pytorch torchvision -c pytorch
git clone https://github.com/Zhcyoung/BEPH_new.git
pip install -U openmim
mim install mmengine
mim install 'mmcv>=2.0.0'
cd mmclassification && mim install -e .
cd .. && cd mmselfsup && mim install -e .
Extract backbone weights to apply to downstream tasks, or download the weight directly [],[]:
import torch
ck = torch.load("./BEPH_weight.pth", map_location=torch.device('cpu'))
outPath = "./BEPH_backbone.pth"
output_dict = dict(state_dict=dict(), author='Yzc')
has_backbone = False
for key, value in ck['state_dict'].items():
if key.startswith('backbone'):
output_dict['state_dict'][key] = value
has_backbone = True
if not has_backbone:
raise Exception('Cannot find a backbone module in the checkpoint.')
torch.save(output_dict, outPath)Downloading + Preprocessing + Organizing TCGA Data
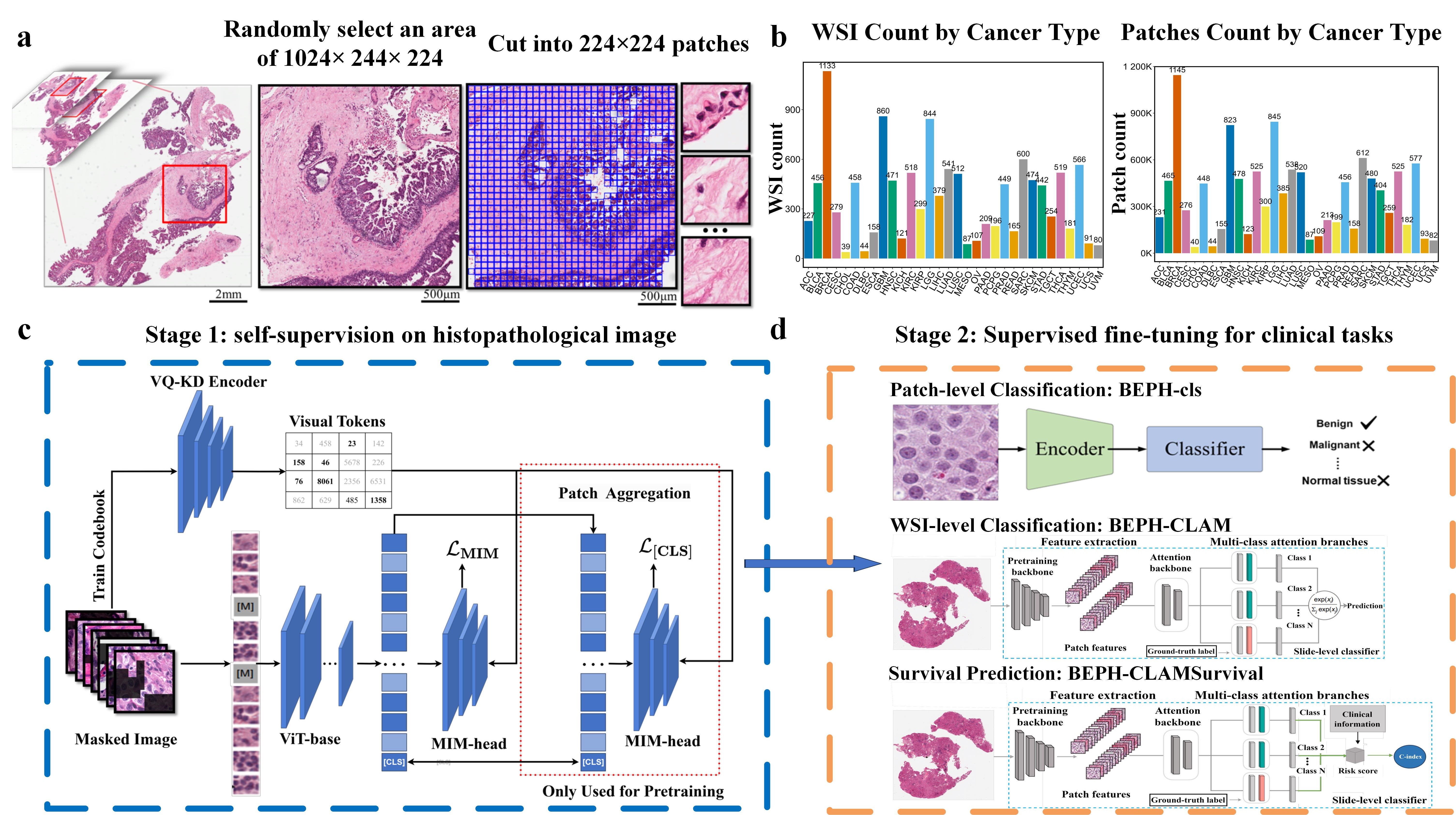
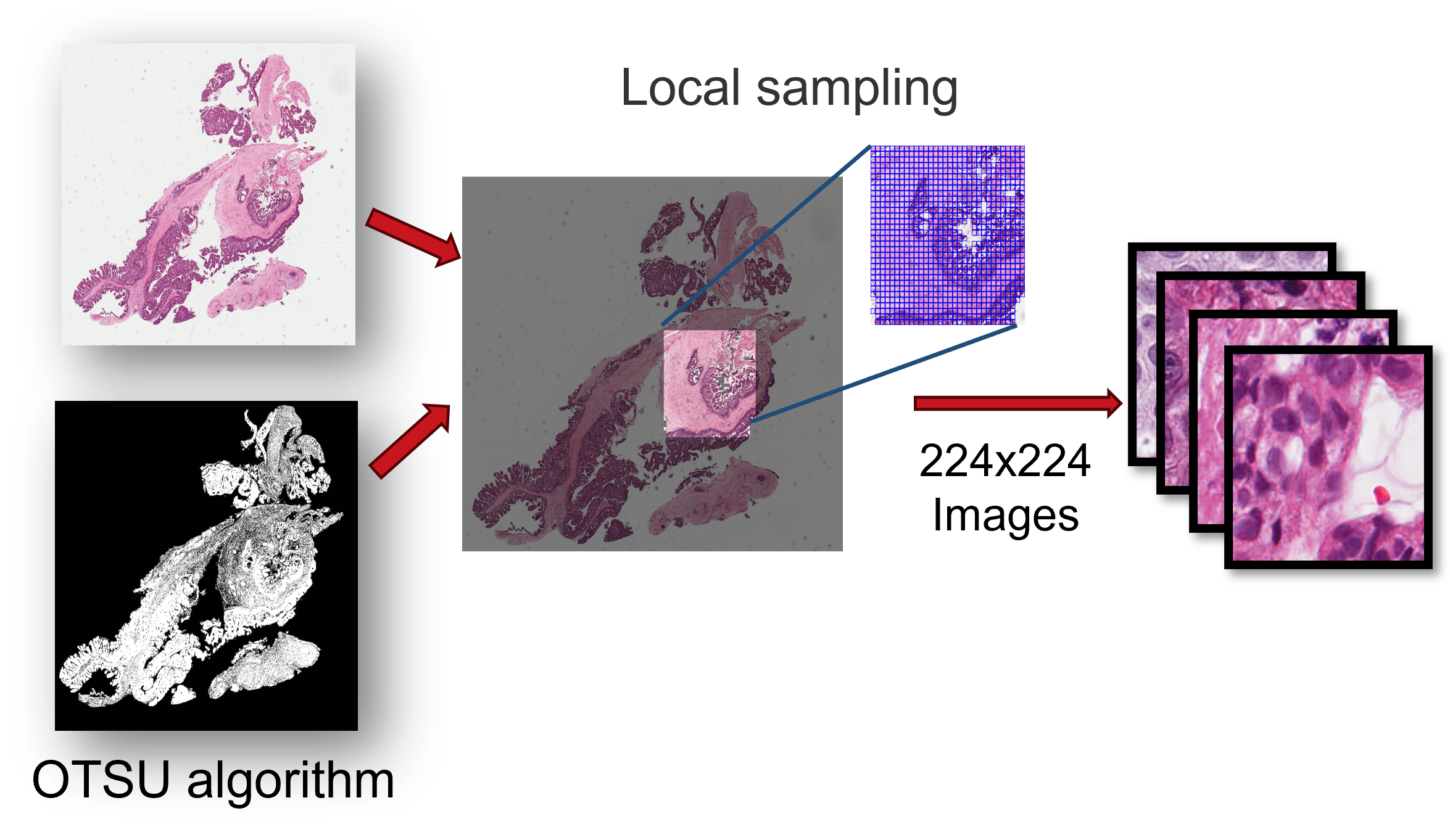
We downloaded diagnostic whole-slide images (WSIs) for 32 cancer types using the GDC Data Transfer Tool, and then we locally sample image regions of 1024×224×224 (approximately 1024 images) from each pathological image, ensuring that the sampled region has a tissue proportion greater than 75%. These sampled image regions are then cropped into 224×224 tiles at 40X magnification, while maintaining a tissue proportion of 75%.
For pre-training,each cancer type is organized as its own folder in TCGA_ROOT_DIR, which additionally contains the following subfolders:
TCGA_ROOT_DIR/
└──tcga_acc/
├── ...
└──tcga_sarc/
├── TCGA-3B-A9HI-01Z-00-DX1
├──0_0.png
├──0_1.png
├──0_2.png
├──...
├── TCGA-DX-A23V-01Z-00-DX1
├── ...
├── ...
├── ...
├── ...And generate a pre-train.txt containing the filename:
./tcga_hnsc/TCGA-CV-A6JZ-01Z-00-DX1/6_30.png
./tcga_dlbc/TCGA-GS-A9TY-01Z-00-DX1/19_15.png
./tcga_gbm/TCGA-06-0171-01Z-00-DX1/27_17.png
... ...And then modify the pre-train config file:beitv2_vit.py
train_dataloader = dict(
batch_size=256,
collate_fn=dict(type='default_collate'),
dataset=dict(
ann_file= 'pre-train.txt' , ###Change to your pre-training file
data_prefix=dict(img_path='Cancer_patches/'),
data_root=data_root,
pipeline=[
dict(type='LoadImageFromFile'),
dict(
brightness=0.4,
contrast=0.4,
hue=0.0,
saturation=0.4,
type='ColorJitter'),
... ...,
)Pre-training Command:
bash tools/slurm_train_4gpu.sh a100 BEPH ./TrainConfigs/beitv2_vit.pyFine-tuning with BEPH weights
To fine tune BEPH on your own data, follow these steps:
- Download the BEPH pre-trained weights, Google Drive, baidu :
Patch level tasks
Start fine-tuning (use BreakHis as example). A fine-tuned checkpoint will be saved during training.
Organise your data into this directory structure:
├── data folder
├──images
├──meta
├──train.txt
├──val.txt
Train.txt /val.txt
./images/SOB_M_DC-14-16716-100-022.png 1
./images/SOB_B_TA-14-16184CD-100-003.png 0
./images/SOB_B_TA-14-16184CD-100-031.png 0
./images/SOB_M_DC-14-14946-100-025.png 1
./images/SOB_M_LC-14-12204-100-037.png 1
... ...Train Command:
bash ./tools/benchmarks/classification/mim_dist_train.sh ./FineTuning/beit.py ./BEPH_backbone.pthFor evaluation (download data and model checkpoints here; change the path below):
bash ./tools/benchmarks/classification/mim_dist_test.sh ./FineTuning/beit.py ./work_dir/epoch_x.pthwsi level tasks:
Following pretraining and pre-extracting instance-level features using ViT-base, we use the publicly-available CLAM scaffold code as well as several of the current weakly-supervised baselines for running 10-fold monte carlo cross-validation experiments.
Directory tree:
DATA_DIRECTORY/
├── slide_1.svs
├── slide_2.svs
└── ...
PATCH_DIRECTORY/
├── masks
├── slide_1.jpg
└── ...
├── patches
├── slide_1.h5
└── ...
├── stitches
├── slide_1.jpg
└── ...
├── process_list_autogen.csv
└── Step_2.csv
FEATURES_DIRECTORY/
├── h5_files
├── slide_1.h5
└── ...
└── pt_files
├── slide_1.pt
└── ...
DATASET_CSV/
└──label.csv
SPLITS/
├── splits_0.csv
└── ...
RESULTS/
├── tcga_brca_subtype
├── s_0_checkpoint.pt
├── splits_0.csv
├── ...
└── summary.csv
└── ...Feature extraction:
python create_patches_fp.py \
--source ./DATA_DIRECTORY/ \
--save_dir ./PATCH_DIRECTORY/patch_splits \
--patch_size 224 \
--seg \
--patch \
--stitch
# --preset tcga.csv \import os
import pandas as pd
df = pd.read_csv('./PATCH_DIRECTORY/process_list_autogen.csv') # This csv is generated in the first step
ids1 = [i[:-4] for i in df.slide_id]
ids2 = [i[:-3] for i in os.listdir('./PATCH_DIRECTORY/patch_splits/patches/')]
df['slide_id'] = ids1
ids = df['slide_id'].isin(ids2)
sum(ids)
df.loc[ids].to_csv('./PATCH_DIRECTORY/patch_splits/Step_2.csv',index=False)
Get feature: histopathological image DINO feature
# ImageNet ResNet-50 feature: extract_features_fp.py
#histopathological image DINO feature: extract_features_dino.py
#BEPH feature: extract_features_BEPH.py
python extract_features_BEPH.py \
--data_h5_dir ./FEATURE_DIRECTORY/patch_splits/ \
--data_slide_dir ./DATA_DIRECTORY/ \
--csv_path ./PATCH_DIRECTORY/patch_splits/Step_2.csv \
--feat_dir ./FEATURES_DIRECTORY \
--batch_size 2000 \
--slide_ext .svs
Filter out the slides that cannot extract features:
df = pd.read_csv(wsi_path[:-3]+'dataset_csv/label.csv')
df = df[['case_id','slide_id','slide_name','oncotree_code']]
ids1 = [i for i in df.slide_name]
ids2 = [i[:-3] for i in os.listdir(wsi_path[:-3]+'test_time_FEATURES_DIRECTORY/pt_files')]
ids = df['slide_name'].isin(ids2)
df = df.loc[ids]
df.columns = ['case_id','slide_id','slide_name','label']
df.to_csv(wsi_path[:-3]+'DATASET_CSV/datasets.csv',index=False)Train Command (Take the clam_sb model for breast cancer subtypes classification as an example):
%run CLAM_SB_BEPH.py \
--data_root_dir DATA_DIRECTORY/ \
--model_type clam_sb \
--task tcga_brca_subtype \
--splits SPLITS/ \
--lr 2e-4 \
--seed 123 \
--feature_path FEATURES_DIRECTORY/
--csv_path DATASET_CSV/datasets.csv \
--k 10 \
--k_start 0 \
--results_dir RESULTS/tcga_brca_subtype
[ "python", "./CLAM_Feature/CLAM_SB_BEPH.py", "--data_root_dir",wsi_path[:-3]+"test_time_FEATURES_DIRECTORY", "--model_type", "clam_sb","--task","Fine_Tuning","--k_start","0","--k",kstart,"--splits",wsi_path[:-3]+ "splits", "--lr", "2e-4", "--seed","47","--csv_path",wsi_path[:-3]+ "/dataset_csv/datasets.csv","--results_dir",wsi_path[:-3]+ str(jobid).split('_')[1]+"/test_result","--early_stopping"]For evaluation:
python eval.py --data_root_dir DATA_DIRECTORY/ \
--model_type clam_sb \
--task tcga_brca_subtype \
--splits SPLITS/ \
--feature_path FEATURES_DIRECTORY/ \
--weights_path ../weights/tcga_brca_subtype/ \
--csv_path DATASET_CSV/label.csv \
--k 10 \
--k_start 0 \
--results_dir RESULTS/tcga_brca_subtype
Analagously, we also extend the CLAM scaffold code for survival prediction, and make available:
Train Command :
python ./survival/CLAM_survival_BEPH.py --data_root_dir DATA_DIRECTORY/ \
--model_type clam_sb \
--task tcga_crc_subtype \
--max_epoch 20 \
--k 5 \
--k_start 0 \
--lr 2e-4 \
--seed 123 \
--results_dir ./RESULTS/tcga_crc_survival\
--early_stopping
# --pretrain_4k vit4k_xs_dino
# 1e-4For evaluation:
python ./survival/eval_survival.py --data_root_dir DATA_DIRECTORY/ \
--model_type clam_sb \
--task tcga_crc_subtype \
--results_dir ./RESULTS/tcga_crc_survival/test