MetDNA2 
About
MetDNA2 excutes knowledge-guided multi-layer metabolic network to annotate metabolites from knowns to unknowns. Generally, the KGMN supports
The KGMN accepts various data imports from common data processing tools, including XCMS, MS-DIAL, and MZmine2. It also support the connection with other metabolomics workflow, like MetFrag, MS-FINDER, MASST etc.
The completed functions are provided in the MetDNA2 webserver via a free registration. The detailed tutorial was also provided in the MetDNA2 webserver.
Installation
You can install MetDNA2 from Github.
if (!require(devtools)){
install.packages("devtools")
}
if (!require(BiocManager)){
install.packages("BiocManager")
}
# Required packages
required_pkgs <- c("dplyr","tidyr","readr", "stringr", "tibble", "purrr",
"ggplot2", "igraph", "pbapply", "Rdisop", "randomForest", "pryr", "BiocParallel", "magrittr", "rmarkdown", "caret")
BiocManager::install(required_pkgs)
# Install ZhuLab related packages
devtools::install_github("ZhuMetLab/SpectraTools")
devtools::install_github("ZhuMetLab/MetBioInterpretation")
# Install `MetDNA2` from GitHub
devtools::install_github("ZhuMetLab/MetDNA2")Note: Due to the limitation of copyright, the library objects zhuMetLib, zhuMetlib_orbitrap, zhuRPlib, lib_rt, lib_ccs are removed in this package. If you want to use the R package, please use your own libray insteaded, and repackage.
Get started
Input
Generally, MetDNA requires the import of the following files for metabolite identifications, including:
- A MS1 peak table (.csv format, required). The first three columns must be "name" , "mz" , and "rt".
- MS2 data files (.mgf or .msp format, required).
- A table for sample information (.csv format, required). The first two columns must be "sample.name" and "group".
- A RT recalibration table (.csv format, optional). If you would like to follow our published LC method and recalibrate the RT library. The gradient of LC are provided here.
The step-by-step tutorials are provided in the MetDNA2 website and the later parts.
Output
The results should be looks like below:
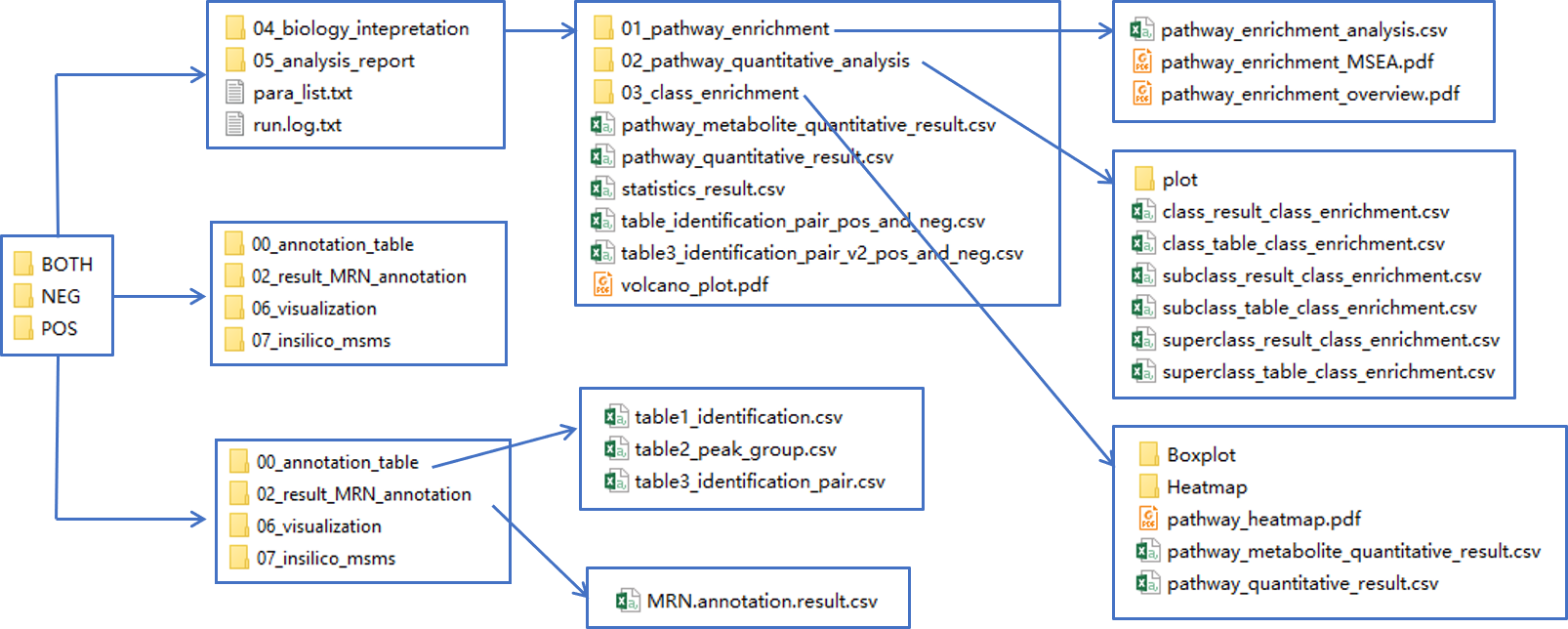
- The
00_annotation_tablecontains annotation results:- The table1_identification.csv contains base peak annotated candidates.
- The table2_peak_group.csv records annotated abiotic peaks in each peak group.
- The table3_identification_pair.csv is same as table 1, but organized as feature-metabolite pairs.
Running on RStudio or R
# load package
library(MetDNA2)
# run MetDNA2
runMetDNA2(
path_pos = "working_directory/POS",
path_neg = "working_directory/NEG",
metdna_version = "version2",
polarity = "positive",
instrument = "SciexTripleTOF",
column = "hilic",
ce = "30",
method_lc = "Other",
correct_p = FALSE,
extension_step = "2",
comp_group = c("W30", "W03"),
species = "hsa",
p_cutoff = 0.050000,
fc_cutoff = 1.000000,
is_rt_calibration = FALSE)Demo data set and Runtime
Generally, it requires 4-8 hours to complete a project, which depends on the number of features and MS/MS spectra. The raw MS data can be found the repository (NIST urine, Fruit fly).
| Project | Running time (hours) | Download | Network |
|---|---|---|---|
| NIST urine (Pos) | 5.4 h | Here | Link |
| NIST urine (Neg) | 8.8 h | Here | Link |
| Head tissue of fruit fly (Pos) | 5.0 h | Here | Link |
| Head tissue of fruit fly (Neg) | 5.9 h | Here | Link |
Connection with other metabolomics workflows
The KGMN is a versatile tool to compatible with various data processing tools and analysis workflow in metabolomics community.
- Note: we provide two packages MetDNA2InSilicoTool and MetDNA2Vis to help user to intergrate with in-silico MS/MS tools and visualize networks, respectively.
| No. | Tool | Usage | Version | Tutorial |
|---|---|---|---|---|
| 1 | XCMS | Peak picking (Input of KGMN) | ≥ v1.46.0 | Tutorial |
| 2 | MS-DIAL | Peak picking (Input of KGMN) | ≥ V4.60 | Tutorial |
| 3 | MZmine | Peak picking (Input of KGMN) | ≥ V3.0.21 | Tutorial |
| 4 | MetFrag | Cross evaluation of KGMN metabolites | ≥ V2.4.5 | Tutorial |
| 5 | CFM-ID | Cross evaluation of KGMN metabolites | ≥ V2.4 | Tutorial |
| 6 | MS-FINDER | Cross evaluation of KGMN metabolites | ≥ V3.24 | Tutorial |
| 7 | MASST | Repository search | ≥ Workflow29 | Tutorial |
| 8 | Cytoscape | Visualization of KGMN | ≥ V5.8.3 | Tutorial |
Need help?
If you have any problems or bug reports, please contact us with the following materials. We will answer your questions at 1:00 pm - 3:00 pm (Beijing time) on every Friday.
- We always welcome any discussions and bug reports about MetDNA via google group: MetDNA forum.
- For Chinese users, please join our QQ group for any discussions and bug reports: 786156544.
Citation
This free open-source software implements academic research by the authors and co-workers. If you use it, please support the project by citing the appropriate journal articles.
Zhiwei Zhou†, Mingdu Luo†, Haosong Zhang, Yandong Yin, Yuping Cai, and Zheng-Jiang Zhu*, Metabolite annotation from knowns to unknowns through knowledge-guided multi-layer metabolic networking, Nature Communications, 2022, 13: 6656 Link
License
 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0)
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0)