MetTracer
Introduction
MetTracer is an R package for tracing isotopic labeling experiments. Current source code still requires some in-house packages, which were not released yet. Hence, users are recommanded to use the docker image we build, which is build with all requirements and easy to use.
The docker image zhulab/mettracer-r contains entire envorienment for running MetTracer. Users can pull it and run MetTracer just as following.
What is MetTracer
MetTracer-r is an Docker environment to processing isotope labelled metabolomics data with MetTracer R package. It is based on the r-base docker.
Pulling image
Users can pull the MetTracer-r image with the following script
docker pull zhulab/mettracer-rData preparation
MetTracer requires the unlabelled metabolomics data to be pre-processed by xcms (see section 3.1.2 in http://metdna.zhulab.cn/metdna/help) and identified by MetDNA (see http://metdna.zhulab.cn) first. If users obtained metabolite identification results via other software, please modify your result table as required
The following files are required by MetTracer for labeled metabolites extraction:
- A folder named
unlabelledcontaining unlabelled data files (.mzXML), annotation result (.csv) and peak information (.Rda) for targe isotopologue generation - A folder named
labelledcontaining data files (.mzXML) for isotope labelling data extraction -
A R script named
run.Rfor your data processingPreparing the labelled folder
The labelled folder contains all the data files requiring extraction of labeled metabolites. We permit multiple subfolders in it, and each subfolder is an independent biological group. The data files in the folder should be in mzXML format.
Preparing the unlabelled folder
The unlabeled folder contains unlabeled data files, annotation result and peak information.
- unlabeled data files
- unlabeled data files are those used for peak detection and metabolite annotation. These data files should be in mzXML format and put into the subfolder.
- annotation information
- annotation information is a .csv format table named
MRN.annotation.result. The table should be put into the subfolder namedMRN_annotation_resultunder subfolderMetProcess-Result
- annotation information is a .csv format table named
- peak information
- peak information is a
.Rdafile which is generated after peak detection withXCMS.
- peak information is a
Note:
if the users use other software tools for metabolite annotation, please adjust the format of annotation table. The annotation table should include columns: name, mz, rt, ID, compound.name, adduct and Formula. If the peak was annotated as several metabolites, separate them with ;

Preparing the R script
File run.R is an R source code file paralleled with labelled folder and unlabelled folder. We provide a template here. Users only need to change the folder name in general. Other parameters are recommended parameters.
- Parameters
adj.labelAll subfolder names inlabelledfolder, the data files in these folder will do isotope contamination correction.adj.unlabelA subfolder inlabelledfolder. The data files in this subfolder are unlabeled, and they will be used as reference files to correct the isotope contamination for data files in alllabelledsubfolder.
wd <- getwd()
setwd(wd)
library(MetTracer)
isotopologueParam <- IsotopologueParam(rt.extend=15, value="maxo")
experimentParam = ExperimentParam(wd = wd, nSlaves = 6)
extractParam <- ExtractParam(d.extract = "labelled",
adj.label = c("Glu_Gln_Ac","Glu_Gln","Gln_Ac","Unlabel"),
adj.unlabel = c(rep("Unlabel", 4)),
adj.contaminate = TRUE)
iso.targets <- GenerateIsotopologue(isotopologueParam, experimentParam)
pdParam <- PeakdetectionParam(peakwidth = c(5,30))
iso.peaks <- ExtractIsotopicPeaks(extractParam, pdParam,
iso.targets,check.peaks = TRUE)
ExtractIsotopologue(extractParam, iso.peaks, iso.targets, correct.iso = TRUE)Overview of the data preparation
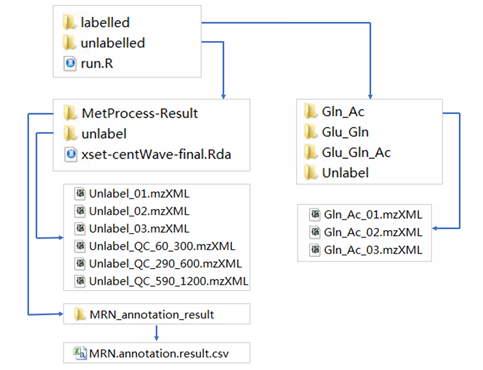
Run data processing work with mettracer-r image
-
go to your data folder (e.g., data)
cd data -
run docker using following code
# MUST keep the code exactly as it is! docker run -it --rm -v "$PWD":/data -u $(id -u ${USER}):$(id -g ${USER}) zhulab/mettracer-r Rscript run.R -
wait till data processing work done
-
Explaining
docker runarguments -
-v "$PWD":/home/${USER}: mapping current dirctory as home directory in docker container -
-u $(id -u ${USER}):$(id -g ${USER}): using current user to run the container -
Rscript ~/run.R: run run.R in container home directory withRscriptcommand
updates
v1.0.1
- bugfix for adduct charge defination
v1.0.4
- update adducts info to match adduct types in MetDNA2
- several minor bugfix
License
 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0)
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0)