Rmagic
true




![]()


Markov Affinity-based Graph Imputation of Cells (MAGIC) is an algorithm for denoising and imputation of single cells applied to single-cell RNA sequencing data, as described in Van Dijk D et al. (2018), Recovering Gene Interactions from Single-Cell Data Using Data Diffusion, Cell https://www.cell.com/cell/abstract/S0092-8674(18)30724-4.
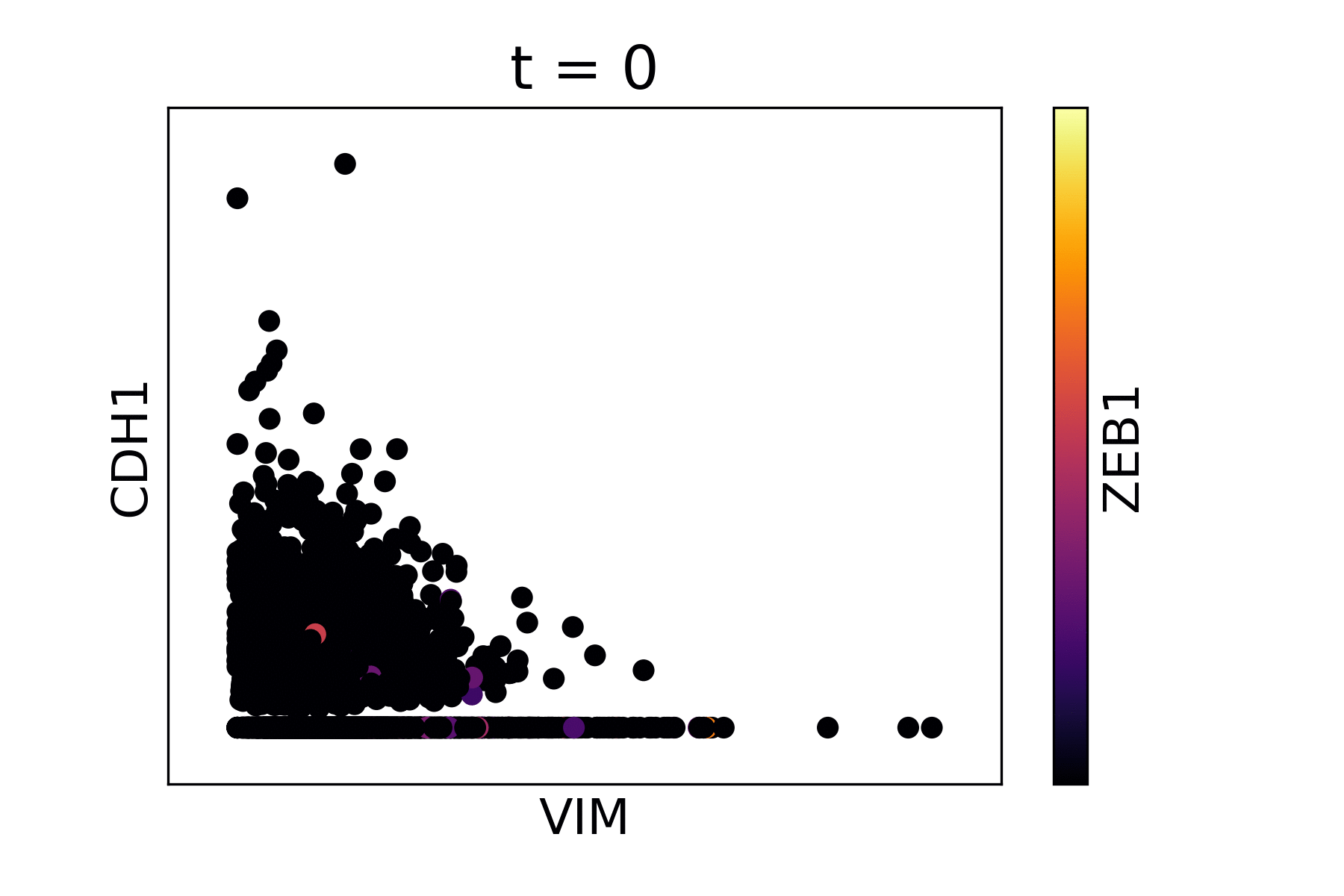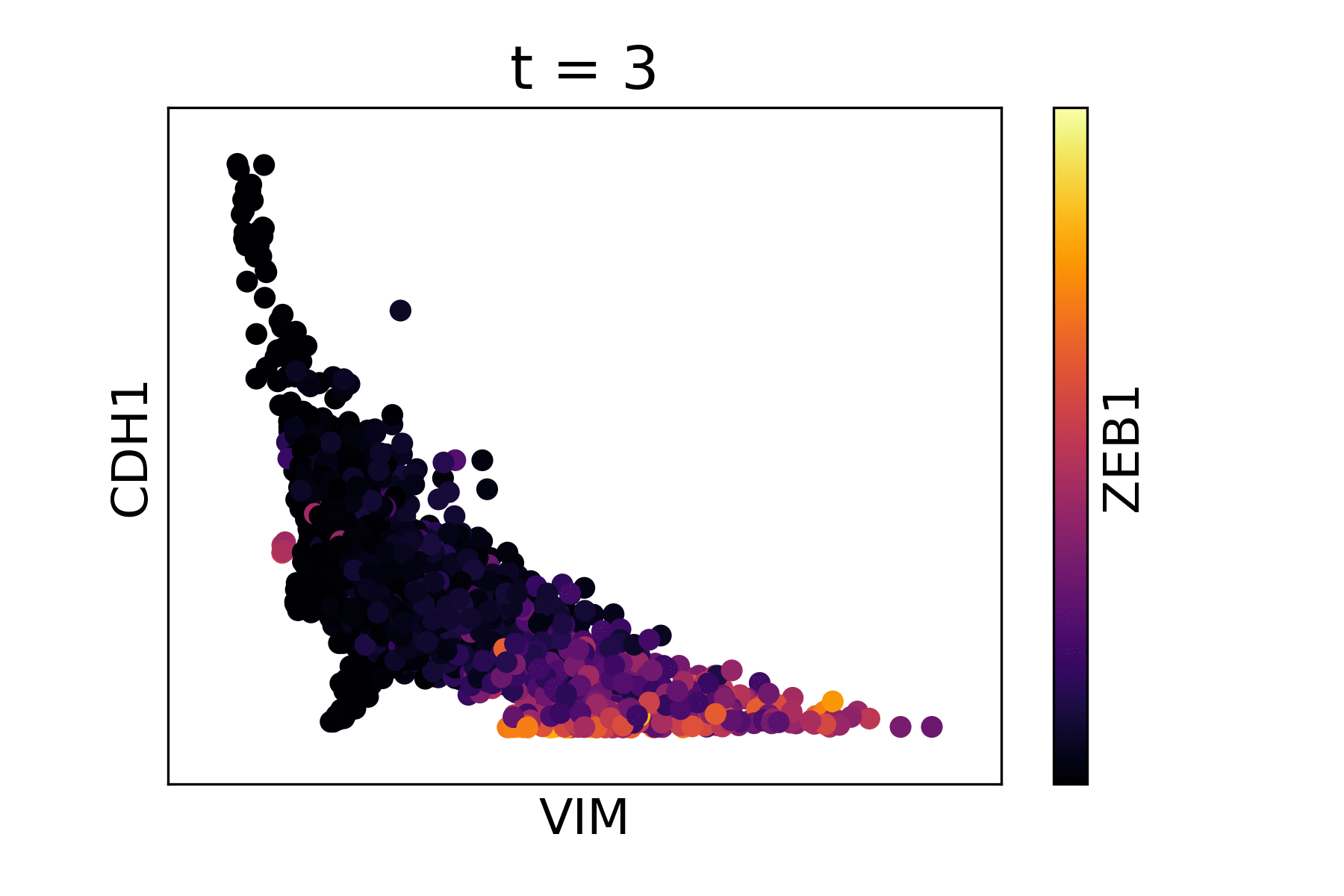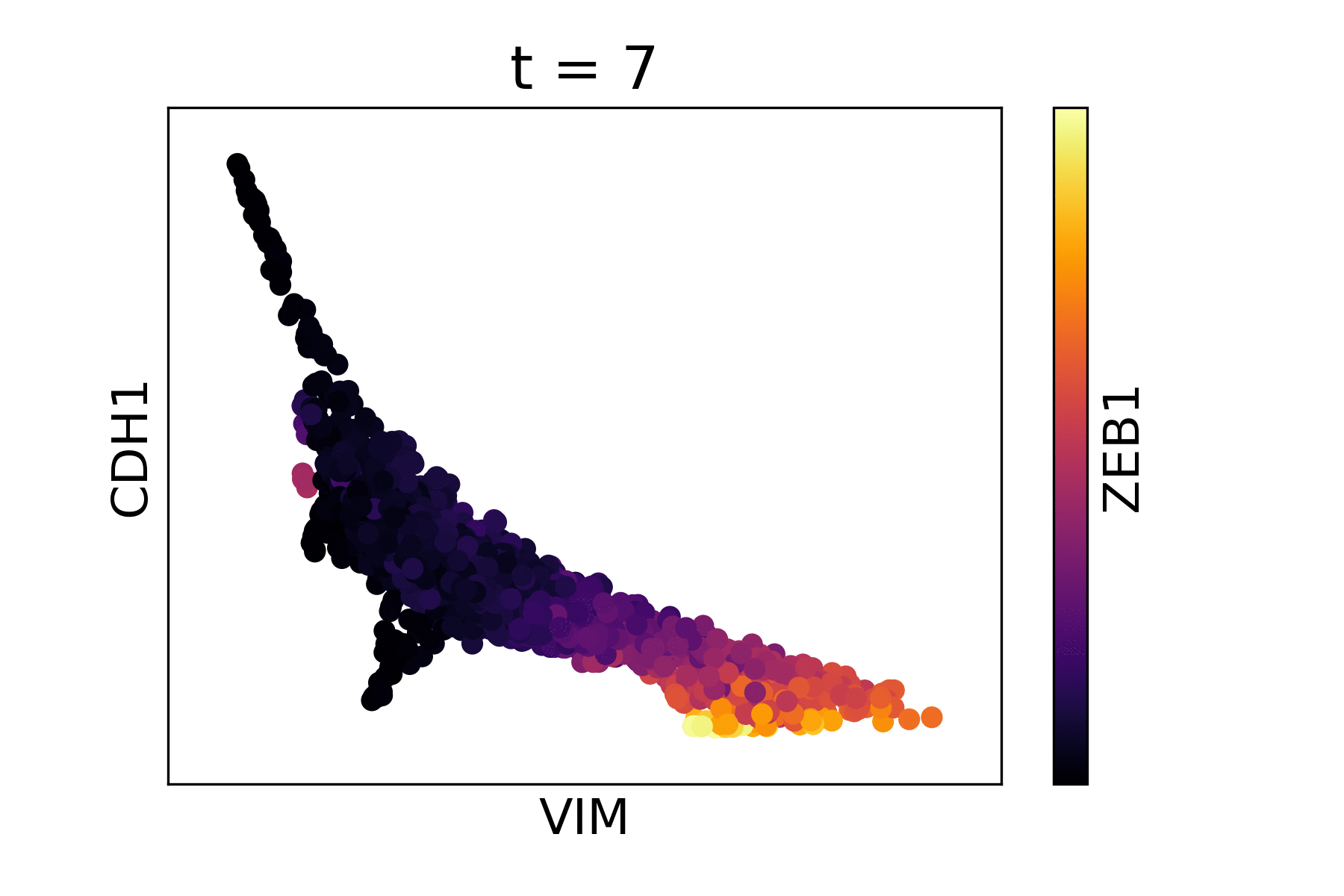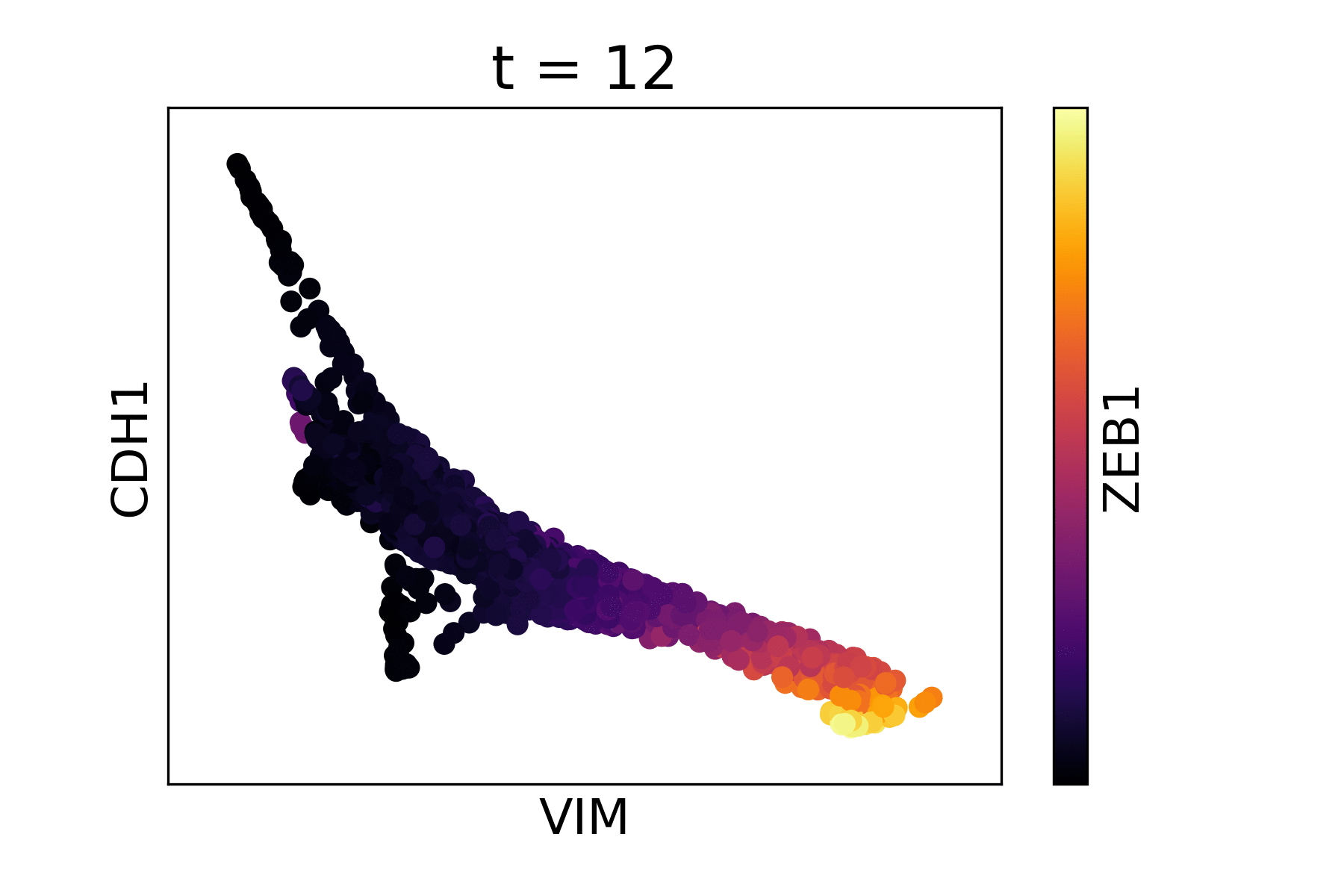
Magic reveals the interaction between Vimentin (VIM), Cadherin-1
(CDH1), and Zinc finger E-box-binding homeobox 1 (ZEB1, encoded by
colors).
- MAGIC imputes missing data values on sparse data sets, restoring the structure of the data
- It also proves dimensionality reduction and gene expression visualizations
- MAGIC can be performed on a variety of datasets
- Here, we show the usage of MAGIC on a toy dataset
- You can view further examples of MAGIC on real data in our notebooks
under
inst/examples:
Table of Contents
Installation
To use MAGIC, you will need to install both the R and Python packages.
If python or pip are not installed, you will need to install them.
We recommend Miniconda3 to install
Python and pip together, or otherwise you can install pip from
https://pip.pypa.io/en/stable/installing/.
Installation from CRAN
In R, run this command to install MAGIC and all dependencies:
install.packages("Rmagic")In a terminal, run the following command to install the Python repository.
pip install --user magic-imputeInstallaton from source
To install the very latest version of MAGIC, you can install from GitHub with the following commands run in a terminal.
git clone https://github.com/KrishnaswamyLab/MAGIC
cd MAGIC/python
python setup.py install --user
cd ../Rmagic
R CMD INSTALL .Quick Start
If you have loaded a data matrix data in R (cells on rows, genes on
columns) you can run PHATE as follows:
library(phateR)
data_phate <- phate(data)Tutorial
Extra packages for the tutorial
We’ll install a couple more tools for this tutorial.
if (!require(viridis)) install.packages("viridis")
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(phateR)) install.packages("phateR")If you have never used PHATE, you should also install PHATE from the command line as follows:
pip install --user phateLoading packages
We load the Rmagic package and a few others for convenience functions.
library(Rmagic)
#> Loading required package: Matrix
library(ggplot2)
#> Warning: package 'ggplot2' was built under R version 3.5.3
library(viridis)
#> Loading required package: viridisLite
library(phateR)
#>
#> Attaching package: 'phateR'
#> The following object is masked from 'package:Rmagic':
#>
#> library.size.normalizeLoading data
The example data is located in the MAGIC R package.
# load data
data(magic_testdata)
magic_testdata[1:5,1:10]
#> A1BG-AS1 AAMDC AAMP AARSD1 ABCA12 ABCG2 ABHD13
#> 6564 0.0000000 0.0000000 0.0000000 0 0 0 0.0000000
#> 3835 0.0000000 0.8714711 0.0000000 0 0 0 0.8714711
#> 6318 0.7739207 0.0000000 0.7739207 0 0 0 0.0000000
#> 3284 0.0000000 0.0000000 0.0000000 0 0 0 0.0000000
#> 1171 0.0000000 0.0000000 0.0000000 0 0 0 0.0000000
#> AC007773.2 AC011998.4 AC013470.6
#> 6564 0 0 0
#> 3835 0 0 0
#> 6318 0 0 0
#> 3284 0 0 0
#> 1171 0 0 0Running MAGIC
Running MAGIC is as simple as running the magic function.
# run MAGIC
data_MAGIC <- magic(magic_testdata, genes=c("VIM", "CDH1", "ZEB1"))We can plot the data before and after MAGIC to visualize the results.
ggplot(magic_testdata) +
geom_point(aes(VIM, CDH1, colour=ZEB1)) +
scale_colour_viridis(option="B")
The data suffers from dropout to the point that we cannot infer anything about the gene-gene relationships.
ggplot(data_MAGIC) +
geom_point(aes(VIM, CDH1, colour=ZEB1)) +
scale_colour_viridis(option="B")
As you can see, the gene-gene relationships are much clearer after MAGIC.
The data is sometimes a little too smooth - we can decrease t from the
automatic value to reduce the amount of diffusion. We pass the original
result to the argument init to avoid recomputing intermediate
steps.
data_MAGIC <- magic(magic_testdata, genes=c("VIM", "CDH1", "ZEB1"), t=6, init=data_MAGIC)
ggplot(data_MAGIC) +
geom_point(aes(VIM, CDH1, colour=ZEB1)) +
scale_colour_viridis(option="B")
We can look at the entire smoothed matrix with genes='all_genes',
passing the original result to the argument init to avoid recomputing
intermediate steps. Note that this matrix may be large and could take up
a lot of
memory.
data_MAGIC <- magic(magic_testdata, genes="all_genes", t=6, init=data_MAGIC)
as.data.frame(data_MAGIC)[1:5, 1:10]
#> A1BG-AS1 AAMDC AAMP AARSD1 ABCA12 ABCG2
#> 6564 0.02565716 0.06303703 0.1726791 0.01559474 0.03114244 0.01423031
#> 3835 0.02535551 0.06286382 0.1678011 0.01547390 0.03017628 0.01428737
#> 6318 0.02619089 0.06298015 0.1744098 0.01514747 0.03145176 0.01477152
#> 3284 0.02517645 0.06254417 0.1684572 0.01559623 0.03015758 0.01414733
#> 1171 0.02651602 0.06289360 0.1729842 0.01514780 0.03162480 0.01480426
#> ABHD13 AC007773.2 AC011998.4 AC013470.6
#> 6564 0.07100262 0.001129400 0.001880153 0.003215547
#> 3835 0.06989726 0.001086716 0.001847604 0.002833342
#> 6318 0.07165035 0.001203505 0.002044504 0.003550067
#> 3284 0.07066602 0.001039065 0.001723499 0.002822357
#> 1171 0.07094679 0.001236082 0.002133401 0.003450875Visualizing MAGIC values on PCA
We can visualize the results of MAGIC on PCA as follows.
data_MAGIC_PCA <- as.data.frame(prcomp(data_MAGIC)$x)
ggplot(data_MAGIC_PCA) +
geom_point(aes(x=PC1, y=PC2, color=data_MAGIC$result$VIM)) +
scale_color_viridis(option="B") +
labs(color="VIM")
Visualizing MAGIC values on PHATE
We can visualize the results of MAGIC on PHATE as follows. We set t
and k manually, because this toy dataset is really too small to make
sense with PHATE; however, the default values work well for single-cell
genomic data.
data_PHATE <- phate(magic_testdata, k=3, t=15)
#> Argument k is deprecated. Using knn instead.
ggplot(data_PHATE) +
geom_point(aes(x=PHATE1, y=PHATE2, color=data_MAGIC$result$VIM)) +
scale_color_viridis(option="B") +
labs(color="VIM")
Issues
FAQ
- Should genes (features) by rows or columns?
To be consistent with common functions such as PCA
(stats::prcomp) and t-SNE (Rtsne::Rtsne), we require that cells
(observations) be rows and genes (features) be columns of your input
data.
- I have installed MAGIC in Python, but Rmagic says it is not installed!
Check your reticulate::py_discover_config("magic") and compare it to
the version of Python in which you installed PHATE (run which python
and which pip in a terminal.) Chances are reticulate can’t find the
right version of Python; you can fix this by adding the following line
to your ~/.Renviron:
PATH=/path/to/my/python
You can read more about Renviron at
https://cran.r-project.org/package=startup/vignettes/startup-intro.html.
Help
Please let us know of any issues at the GitHub
repository. If you
have any questions or require assistance using MAGIC, please read the
documentation by running help(Rmagic::magic) or contact us at
https://krishnaswamylab.org/get-help.