 gwaybio
commented
2 years ago
gwaybio
commented
2 years ago Glad to hear you've found our tool useful @rjesud !
samples is a flexible argument that must be input in the format of a pandas.DataFrame.query(). All it is doing is learning a specific transform (e.g. zscore) for only the samples provided in the query and then applying this transform to the full data. So, for example, you might want to use the argument if you do, as you say, are combining data from multiple plates that contain the same controls but different treatments.
spherize doesn't "require" the samples argument - it should work perfectly fine with the default. However, in practice, you probably should use the samples argument. You can find a direct example of our pipeline here: https://github.com/broadinstitute/lincs-cell-painting/. You'll probably derive the most benefit from the profiles and spherized_profiles folders. You'll notice that we did apply spherize afer the first round of normalization and feature selection (see here), but this is likely data-specific and a case-by-case basis.
I am also generally very interested in how you're using the package. We are thinking about writing this paper up, and it might be nice to discuss potential use cases. Additionally, it would be great to highlight other, community examples by providing links to examples somewhere in this repo!
 rjesud
rjesud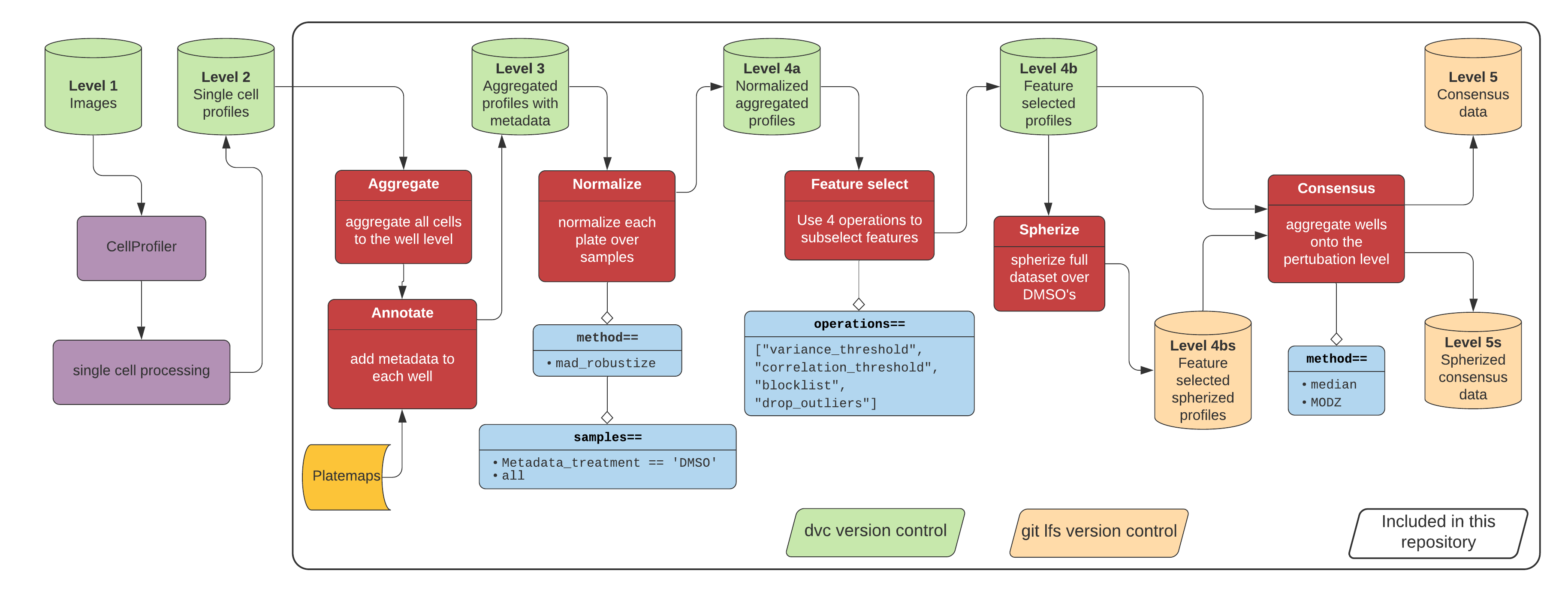{kind=link}
Firstly, thanks for releasing this tool. It has been a great resource for our projects.
I am looking for clarification on the normalization function - namely, when is it appropriate to use/skip the "samples" argument and how exactly is the normalization method using this flagged data. Is this only necessary when combining data from multiple plates (each with control/dmso wells)?
Secondly, is it correct to say that the 'spherize' method requires the 'samples' argument? And this should be executed AFTER a first round normalization and feature selection?
Thank you!